
| File Formats |


This book lists only the file formats that are used by more than one Insight II product or module. You should always look in the File Formats appendix of each product's user guide for information about formats that are unique to that product.
The appendix of this book describes fixed, non-changing formats that were used by earlier versions of Insight II software and that can still be used. Each of these classic formats has a current version described in this main section of the book.
Table 1 lists the products or modules that use the formats described in this book.
An alignment of two or more amino acid sequences can be read into Consensus or Homology from a .align file with the Get Sequences Alignment command. The filename should end in the extension .align, since only files with this extension are listed in the value-aid in the Get Sequences Alignment command. The file is a text file containing lines of no more than 1000 characters each. Shorter lines (typically 80 characters or less) can be used, if desired, to make the file easier to read and edit. Each line begins with a protein name (up to six characters long) followed by a colon (":"). The remainder of the line contains the amino acid sequence of the named protein. In addition to the single-letter amino acid codes listed above, the sequence may contain gap characters ("-") and break characters ("|"); the latter indicate breaks between protein chains. The text lines are organized into blocks of lines, each block containing exactly one line for each protein in the alignment. Blocks are separated from one another by one or more blank lines; therefore no blank lines are allowed within a block. The proteins must be listed in the same order in all blocks in the file.
Sequence Alignment Files (.align)
Sample .align File
Here is an example of a sequence alignment in the correct format for an alignment file:
# Comments like this one are completely ignored.
! The exclamation mark also denotes a comment.
# Note how leading space characters are used in the first
# block to establish the proper alignment of the N- termini of
# the four sequences. Also notice that a "place-holding" line
# is required in the third block because the short sequence IER
# does not extend that far. IER: mtqspsslsas-vgdrvtitcqas------qdiikylnwyqqtpgka
PCM1: VMTQSPSSLSVSA-GERVTMSCKSSQSLLNSGNQKNFLAWYQQKPGQP
JBF: EIVLTQSPAITAASL-GQKVTITCSASSS-------VSSLHWYQQKSGTS
F91: IQMTQT-TSSLSASLGDRVTISCRASQD------ISNYLNWYQQKPDGT IER: pklliyeasnlqagvpsrfsgsgsgtdytftisslqped PCM1: PKLLIYGASTRESGVPDRFTGSGSGTDFTLTISSVQAEDLAVYYCQN DHS
JBF: PKPWIYEISKLASGVPARFSGSGSGTSYSLTINTMEAEDAAIYYCQQWT-
F91: VKLLVYYTSRLHSGVPSRFSGSGSGTDYSLTISNLEHEDIATYFCQQGST IER: # place holder
PCM1: YP-LTFGAGTKLEIKRADAAPTVSIFPPSSEQLTSGGAS
JBF: YPLITFGAGTKLELKRADAAPTVSIFPPSSEQ
F91: TP-RTFGGGTKLEIKRRADAAPTVSIFPPS
The format is:
HELIX header line (if present)
PBC header line
title/energy line
date line
PBC record (if present)
HELIX record for molecule A (if present)
atom record for atom 1
.
.
.
atom record for atoms i
end
HELIX record for molecule B (if present)
atom record for atom i + 1
.
.
.
atom record for atoms j
end
HELIX record for molecule C (if present)
atom record for atom j + 1
.
.
.
atom record for atoms k
end
.
.
.
end
end
title/energy line
date line
PBC record (if present)
HELIX record for molecule A (if present)
atom record for atom 1
.
.
.
atom record for atoms i
end
HELIX record for molecule B (if present)
atom record for atom i + 1
.
.
.
atom record for atoms j
end
HELIX record for molecule C (if present)
atom record for atom j + 1
.
.
.
atom record for atoms k
end
.
.
.
end
end
Sample .arc File
The example shows an .arc file for a 2D-periodic, helical system with three frames. Ordinarily, an .arc file consists of a single long line with no carriage returns. Carriage returns have been inserted into the following example to make it more understandable.
!BIOSYM archive 3
HELIX
PBC=2D
Frame 1
!DATE Thu Jul 19 18:39:47 1993
PBC 18.6200 18.6200 90.0000 (P 1)
HELIX 143.3598 7.6194 90.0000 90.0000 0.0000 0.0000
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c3 C -0.300
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
HELIX 121.2043 17.6194 90.0000 90.0000 0.5000 0.4500
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
F11 11.762401581 -1.450428009 -9.612532616 ETHE 1 f F 0.100
H11 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
Frame 2
!DATE Thu Jul 19 18:39:47 1993
PBC 18.6200 18.6200 90.0000 (P 1)
HELIX 143.3598 7.6194 90.0000 90.0000 0.0000 0.0000
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c3 C -0.300
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
HELIX 121.2043 17.6194 90.0000 90.0000 0.5000 0.4500
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
F11 11.762401581 -1.450428009 -9.612532616 ETHE 1 f F 0.100
H11 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
Frame 3
!DATE Thu Jul 19 18:39:47 1993
PBC 18.6200 18.6200 90.0000 (P 1)
HELIX 143.3598 7.6194 90.0000 90.0000 0.0000 0.0000
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c3 C -0.300
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
HELIX 121.2043 17.6194 90.0000 90.0000 0.5000 0.4500
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
F11 11.762401581 -1.450428009 -9.612532616 ETHE 1 f F 0.100
H11 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
Note that .car files are used by several Insight II products; therefore, some of the information present may be ignored by some programs or used only by certain programs. In particular, the ability of the .car file to support helical and 2D periodic systems is accomplished by the presence of special lines that are present only if the .car file contains 2D periodic and/or helix information. This information is currently relevant only to the Polymer product, and in that context, only to the Discover 97.0/3.0.0 program. In addition, since the Discover program does not handle infinite helices, it does not read .car files containing helix information. If a .car file contains 2D periodicity without helix information, however, the Discover 97.0/3.0.0 program (but not the Discover 2.9.x program) can read it and can also write files for these systems.
There are several differences between the file format described in the Discover 2.9.0 and Insight 2.2.0 documentation and the format presented here, to enable atom names, potential types, and residue names to be longer than in previous versions.
The .car file consists of one file header (which includes statements indicating what kinds of information are present in the file), one coordinate section header, optional 2D or 3D periodicity records, one coordinate section for each molecule in the file, and one end-of-file statement. All lines in the file are exactly 80 characters long. The coordinate section(s) includes:
File Header
The first record of a .car file must be:
!BIOSYM archive #The ! must be the first character in the file. The Discover program interprets this line as indicating an ASCII file containing coordinate records as outlined in this section. The string archive indicates that the contents of the file are those of a .car file; the # identifies the file format. For example, 3 indicates that the file format is as specified here for the Discover program, versions 2.9.5/3.2 and later.
If helix information is not present in the .car file, then the second line indicates whether the file contains PBC information. If helix information is present, then the second line of the .car file consists of the word HELIX and the third line indicates whether the file contains PBC information. Note that helical symmetry is not currently compatible with 3D periodicity. So if the second line is HELIX and PBC=ON is found, an error message is generated.
Valid file headers:
!BIOSYM archive 3
HELIX
PBC=2D !BIOSYM archive 3
HELIX
PBC=OFF !BIOSYM archive 3
PBC=ON !BIOSYM archive 3
PBC=OFF !BIOSYM archive 3
PBC=2D
Periodicity Records
A periodicity record is present in the .car file only if the entry PBC=ON or PBC=2D is present in the file header.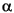 ,
, 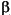 , and
, and  angles, and the space group name. Please see the Discover 2.9.x/97.0/3.0.0 User Guide for a discussion of valid space group names.
angles, and the space group name. Please see the Discover 2.9.x/97.0/3.0.0 User Guide for a discussion of valid space group names.
For 2D periodicity, indicated by the file header line PBC=2D, the PBC section (Tables 2 and 2) contains the word PBC, the k and l values, the value of , and the plane group name. These coordinates are explained in the Insight and Polymer documentation. Currently, only the (P1) plane group is supported, and only the Discover 97.0/3.0.0 program (not the Discover 2.9.x program) reads such files.
Example PBC record for 2D periodicity:
PBC 18.6200 18.6200 90.0000 (P 1)
If a molecule has helical symmetry (i.e., it is an infinite helix), then an extra line is present in the relevant coordinate section, just before the atom records for that molecule. Note that each helical molecule has its own helix record. The new line in the coordinate section contains the word HELIX, followed by the
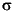 and d values, the
and d values, the 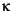 and
and 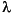 angles, and the Tk and Tl positions (see Table 2). These coordinates are explained in the Insight and Polymer documentation.
angles, and the Tk and Tl positions (see Table 2). These coordinates are explained in the Insight and Polymer documentation.HELIX 143.3598 7.6194 90.0000 90.0000 0.5000 0.4500
End Records
The coordinate section for each molecule in the system must end with the word "end" in the first three columns of the last line of the section. In addition, the entire file must end with the word "end", also in the first three columns of the last line of the file.
Sample .car Files
The following examples indicate the correct format for .car files (although the data are not necessarily physical).
Example 1: Nonperiodic, Nonhelical System
1 2 3 4 5 6 7 8
12345678901234567890123456789012345678901234567890123456789012345678901234567890
!BIOSYM archive 3
PBC=OFF
!DATE Thu Jul 19 18:39:47 1993
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c2 C -0.200
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
H11 11.762401581 -1.450428009 -9.612532616 ETHE 1 h H 0.100
H12 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
!BIOSYM archive 3
PBC=ON
!DATE Thu Jul 19 18:39:47 1993
PBC 18.6200 18.6200 18.6200 90.0000 90.0000 90.0000 (P 1)
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c2 C -0.200
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
H11 11.762401581 -1.450428009 -9.612532616 ETHE 1 h H 0.100
H12 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
1 2 3 4 5 6 7 8
!BIOSYM archive 3
PBC=2D
!DATE Thu Jul 19 18:39:47 1993
PBC 18.6200 18.6200 90.0000 (P 1)
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c2 C -0.200
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
H11 11.762401581 -1.450428009 -9.612532616 ETHE 1 h H 0.100
H12 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
!BIOSYM archive 3
HELIX
PBC=OFF
!DATE Thu Jul 19 18:39:47 1993
HELIX 143.3598 7.6194 90.0000 90.0000 0.0000 0.0000
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c2 C -0.200
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
HELIX 121.2043 17.6194 90.0000 90.0000 0.0000 0.0000
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
H11 11.762401581 -1.450428009 -9.612532616 ETHE 1 h H 0.100
H12 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
1 2 3 4 5 6 7 8
!BIOSYM archive 3
HELIX
PBC=2D
!DATE Thu Jul 19 18:39:47 1993
PBC 18.6200 18.6200 90.0000 (P 1)
HELIX 143.3598 7.6194 90.0000 90.0000 0.0000 0.0000
C1 3.108016491 0.653186858 -8.526236534 ETHE 1 c2 C -0.200
H11 2.814816952 -0.348720580 -8.761003494 ETHE 1 h H 0.100
H12 2.517393827 1.015321732 -7.710808277 ETHE 1 h H 0.100
C2 4.596541882 0.674196541 -8.131963730 ETHE 1 c2 C -0.200
H21 4.748424053 0.049042296 -7.276969910 ETHE 1 h H 0.100
H22 4.889741421 1.676103950 -7.897196770 ETHE 1 h H 0.100
end
HELIX 121.2043 17.6194 90.0000 90.0000 0.5000 0.4500
C1 11.610512733 -0.825278699 -10.467529297 ETHE 1 c2 C -0.200
H11 11.762401581 -1.450428009 -9.612532616 ETHE 1 h H 0.100
H12 11.903706551 0.176631495 -10.232768059 ETHE 1 h H 0.100
C2 12.459976196 -1.346121073 -11.640320778 ETHE 1 c2 C -0.200
H21 12.166784286 -2.348031998 -11.875079155 ETHE 1 h H 0.100
H22 12.308086395 -0.720973670 -12.495317459 ETHE 1 h H 0.100
end
end
If you want to continue a minimization from the point at which a previous minimization finished, then rename the output .cor file to an input .car file before performing the minimization. Alternatively, the .cor file name can be specified in the begin command (see the Discover 2.9.x/97.0/3.0.0 User Guide for versions 2.9.x and 97.0/3.0.0, respectively, of the Discover program).
The format of the .cor file is identical to that of the .car file.
There are three kinds of record types in the element data file:
Example:
# code vdw radius cov. radius min val max val common weight
element H 1.10 0.32 1.0 1.0 1.0 1.008
element C 1.55 0.77 4.0 4.5 4.0 12.011
element N 1.40 0.75 2.5 5.0 3.0 14.007
| Contents | Comment |
|---|---|
| bond | Record identifier |
| element code | One- or two-letter element name |
| element code | One- or two-letter element name |
| bond length | Bond length in angstroms |
bond H N 1.03
bond C C 1.54
bond C O 1.43
bond L S 1.1
Format files consist of sequentially executed commands from the set described below. All but the FORMAT and BOND_TABLE commands are a single line. The commands in the group beginning FORMAT_ are followed by any number of field specifiers and terminated by an END_FORMAT command. All commands and field specifiers must appear exactly as listed here; no abbreviations or lower case letters are allowed.
IGNORE_FOR number
On input: Skip the given number of lines. Useful for skipping a fixed length header section (a line of the header can be read as the TITLE field).
On output: No function on output.
IGNORE_TO [start] string
On input: Skip lines until a line is encountered which contains the given string. The optional start parameter may be used to indicate in what column testing for the match should begin, or may be * to check for a match anywhere in the line. Note that column numbering begins at 1 for the leftmost column. If no start column is supplied, matching begins in column 1. This command might be used to get down to the ATOM section of a pdb file.
On output: No function on output.
IGNORE_WHILE [start] string
On input: Skip lines while they match the string given. The optional start parameter functions as in IGNORE_TO.
On output: No function on output.
MARKER [start] string
On input: Skip one line from the input file
On output: Output the given string starting in the column specified or column 1 if no start is given. An asterisk (*) given for the start column is interpreted as column 1 in this command. Used for such things as END markers that separate atom and connectivity sections of the file.
FORMAT_FOR number
On input: Read the specified number of lines using the format that follows. The number field is often a symbolic variable such as $NUM_ATOMS filled by an earlier read.
On output: Write the specified number of lines using the given format. When fields corresponding to atom data are included, the data come from the list of specified atoms starting at the beginning and advancing one atom each time the format is applied. If a symbolic variable like $NUM_ATOMS is used, then it is evaluated to the number of atoms in the object specified in the put command.
FORMAT_TO [start] string
On input: Read input lines using the format until a line containing the given string is encountered. The line with the matching string is not processed. This type of read can be used in conjunction with the MARKER command for files with sections separated by markers such as END.
On output: Write out the information in the format for every atom specified in the put command.
FORMAT_WHILE [start] string
On input Read input lines until encountering a line whose initial characters do not match the given string. Stops so that this non-matching line is the next line to be read. This type of read is designed for pdb-style files where sections are delimited by different keywords. The length and presence of the string does not affect the columns of the format specification.
On output: Write out the information in the format for every atom in the list. The string does not automatically appear in the line being written but may be output using a MARKER field in the format.
FORMAT_TO_EOF
On input: Read input lines using the format until the end of the input file is encountered. This type of read should be the last in a format description file, since all subsequent reads will fail.
On output: Write out the information in the format for every atom in the list.
END_FORMAT
On input: Marks end of a format specification.
On output: Marks end of a format specification.
BIDIRECTIONAL_BONDS
On input: Lets the system know that all bonds are listed twice, once in each direction. Insight .mdf files use this convention.
On output: Lets the system know that all bonds are listed twice, once in each direction. Insight .mdf files use this convention.
The regular field types are described in Table 6.
Format Descriptions
A format description consists of one of the format commands, followed by a variable number of field definitions and terminated by an END_FORMAT command. When processing a format there is a notion of current position which is important for delimited reading. The initial position is at the start of the input line. As the program reads each of the defined fields, it starts at either the current position, if the start of the field is *, or moves to the given column if one is supplied. If the field length is *, then reading continues until a delimiter is encountered (if a non-digit is found in an integer or float field, reading stops there as if it were a delimiter). If the field length is explicitly given then reading continues for exactly that many characters or until end of line, whichever happens first.
Bond Code Table
To accommodate a wide variety of bond order representations, the free format utility allows definition of a bond code table. This table allows you to associate the bond order codes of the file format being read/written with the Insight II bond orders. A table giving the code for any or all of the Insight II bond orders (SINGLE, DOUBLE, TRIPLE, PART_DOUBLE) is given in the following example:
BOND_TABLEWhen using this example during input, a BOND_ORDER field with the value of 1.5 is interpreted as a partial double bond. On output the BOND_ORDER for a triple bond is written as 4.
PART_DOUBLE 1.5
TRIPLE 4
END_TABLE
If no bond table is given, the default bond order codes are:
1=SINGLE
2=DOUBLE
3=TRIPLE
4=PART_DOUBLE
Important in this system is the notion of current atom. When you begin processing a file there are no atoms defined and hence the current atom is null. After processing a format in which atoms were defined, the atom list is non-empty. Before starting to process a subsequent format, set the current atom to the first atom of the atom list. Then after every application of the format you advance the current atom to the next in the list. This automatic stepping down the atom list provides for an implicit correspondence between different sections of an input file. The most common example is a file that has an atom definition section, and then a connectivity section where the lines correspond sequentially to the atoms in the atom section. When atom names or numbers are explicitly specified, an attempt is made to find that atom in the existing atom list and make it the current atom.
These rules are:
If there is a DEFINE_ATOM in the format
{
create a new atom and make it the current atom
}
else if there is an atom number/name in format
{
find the specified atom and make it the current atom
}
Add the fields read to the current atom
Advance the current atom to next in the list
# SYBLIKE.FRM
# Format file for doing free_format input/output of syblike files
#
IGNORE_WHILE "*" # interpret bond order of 5 as partial double
BOND_TABLE
PART_DOUBLE 5
END_TABLE # number of atom records
FORMAT_FOR 1
NUM_ATOMS 1 4
MARKER 6 "MOL"
TITLE 12 100
END_FORMAT # atom records
FORMAT_FOR $NUM_ATOMS
DEFINE_ATOM
ATOM_NUMBER 1 4
ELEMENT_NUMBER 5 4
ATOM_X 9 9
ATOM_Y 18 9
ATOM_Z 27 9
ATOM_NAME 36 4
END_FORMAT IGNORE_WHILE "*" # number of bond records
FORMAT_FOR 1
NUM_BONDS 1 4
MARKER 6 "MOL"
END_FORMAT # bond records
FORMAT_FOR $NUM_BONDS
BOND_NUMBER * 4
BOND_FROM_NUMBER * 4
BOND_TO_NUMBER * 4
SPACES 9
BOND_ORDER * 4
END_FORMAT MARKER "0 MOL"
# CHEMDLIKE.FRM
# chemdlike format file for free format input/output
#
BIDIRECTIONAL_BONDS
LINE_LENGTH 85 IGNORE_WHILE "*" # cell parameters
FORMAT_FOR 1
CELL_A 39 8
CELL_B 47 8
CELL_C 55 8
END_FORMAT
FORMAT_FOR 1
ALPHA 22 8
BETA 30 8
GAMMA 38 8
END_FORMAT # number of atoms
FORMAT_FOR 1
NUM_ATOMS 1 4
TITLE 10 60
END_FORMAT MARKER " Title2 not used"
FORMAT_FOR $NUM_ATOMS
DEFINE_ATOM
# special marker string to put 0's in all bond_to fields that
# will not be filled with actual bonds
MARKER 42 " 0 0 0 0 0 0 0 0"
ATOM_NUMBER 1 4
ATOM_NAME 6 4
ATOM_X 12 9
ATOM_Y 22 9
ATOM_Z 32 9
BOND_TO_NUMBER 42 4
BOND_TO_NUMBER * 4
BOND_TO_NUMBER * 4
BOND_TO_NUMBER * 4
BOND_TO_NUMBER * 4
BOND_TO_NUMBER * 4
BOND_TO_NUMBER * 4
BOND_TO_NUMBER * 4
CHARGE 75 7.3
# marker for atom group field
MARKER 85 "1"
END_FORMAT
# PDBLIKE.FRM Revised 7/13/89
# Format file for doing free format input/output of pdblike files
#
# NOTE: Since the connectivity section may contain lines with
# fewer bonds than the possible four, there may be messages about
# inability to find atoms to connect to. IGNORE_TO "ATOM" FORMAT_WHILE "ATOM"
DEFINE_ATOM
MARKER 1 "ATOM"
ATOM_NUMBER 7 5
ATOM_NAME 14 3
RESIDUE_TYPE 18 3
RESIDUE_NUMBER 23 4
ATOM_X 31 8.3
ATOM_Y * 8.3
ATOM_Z * 8.3
END_FORMAT MARKER "TER" # PDB files specify bonds twice, once in each direction, so we
# need to set the bidirectional bonds flag
BIDIRECTIONAL_BONDS FORMAT_WHILE "CONECT"
MARKER 1 "CONECT"
BOND_FROM_NUMBER 7 5
BOND_TO_NUMBER * 5
BOND_TO_NUMBER * 5
BOND_TO_NUMBER * 5
BOND_TO_NUMBER * 5
END_FORMAT
# MDLLIKE.FRM
# Format file for doing free format input/output of mdllike files FORMAT_FOR 1
TITLE 1 80
END_FORMAT #Molecule Header
MARKER "" #Comments
MARKER "File Written using Insight Free Format Output" #number of atoms and bonds
FORMAT_FOR 1
NUM_ATOMS 1 3
NUM_BONDS * 3
END_FORMAT #atom records
FORMAT_FOR $NUM_ATOMS
DEFINE_ATOM
ATOM_X * 10.4
ATOM_Y * 10.4
ATOM_Z * 10.4
SPACES 1
ELEMENT_NAME * 3
#NOTE: we cannot do the charges because they are coded in a non-
#standard way
END_FORMAT; #bond records
FORMAT_FOR $NUM_BONDS
BOND_FROM_NUMBER * 3
BOND_TO_NUMBER * 3
BOND_ORDER * 3.0
END_FORMAT
As with graph files, you may include any number of comment lines at the top of the file. You may define as many graphs as you like, but remember that only nine graphs fit on the screen without overlapping one another.
Each graph may define multiple plots and may define the title of the graph. Each plot may define the color to be used, the point connection, and the symbol to use if points are to be displayed.
Each element of the graph definition is identified by a string. GRAPH indicates a new graph. The string TITLE is optional. If you want to give the graph a title, enter TITLE on the line following GRAPH. Follow TITLE with a space and then the actual title.
PLOT indicates a new plot. As mentioned above, a graph definition may contain several plot definitions. For each plot you may optionally specify:
Following the optional display definitions, the x, y, and optionally z, functions are defined using the keywords X FUNCTION, Y FUNCTION, and Z FUNCTION, followed by the name of the function for that axis.
Below is the order in which each graph element definition should occur and which elements are optional:
GRAPH <required>
Blank lines may occur only in the comments at the top. Graph and plot definitions may not contain or be separated by any blank lines. If any required elements are missing or in the wrong order an error declaring a bad file format is displayed.
For standard graphs, all functions given in the .grf must be contained within the graph data file (.tbl). If a specific function in the standard graph definition file (.grf) cannot be located in the graph data file (.tbl), an error does not occur, but an informational message is displayed and the plot is not created.
The first is 2D, with only one plot and a title. Notice that you may optionally specify a Z Function.
The second is 3D, with only one plot. This graph definition accepts the default color and point connection attributes.
The third graph defines two plots, and each is 2D. The first plot defines RED to be the color. The second plot defines the color to be BLUE (RED and BLUE are hues; see the Graph/Color commands description), and specifies that only the points should be displayed (CONNECTION is OFF) using the TRIANGLE symbol.
The next graph defines two plots, the first 3D and the second 2D. In the first, only points are displayed using the BOX symbol. The color uses an RGB specification; in this case yellow. In the second plot, both lines and points are displayed (if not specifically turned off, CONNECTION is ON). The color is light blue, the point symbol is a STAR, the scale of the points is 4.0.
The last graph defines a single 2D plot. The color is yellow, with a bar display, and Y specified as the independent axis.
GRAPH ! First graph !
TITLE Sample 1
PLOT ! Only plot in first graph !
X FUNCTION function_1
Y FUNCTION function_2
GRAPH ! Second graph !
PLOT ! Only plot in second graph!
X FUNCTION time
Y FUNCTION energy
Z FUNCTION None
GRAPH ! Third graph !
PLOT ! First plot in third graph !
COLOR RED
X FUNCTION function_a
Y FUNCTION function_b
PLOT ! Second plot in third graph !
COLOR BLUE
CONNECTION off
POINT SYMBOL TRIANGLE
X FUNCTION function_c
Y FUNCTION function_c
GRAPH ! Fourth graph !
TITLE Sample
PLOT ! First plot in fourth graph !
COLOR 255,255,0
CONNECTION OFF
POINT SYMBOL BOX
X FUNCTION function_a
Y FUNCTION function_b
Z FUNCTION function_c
PLOT ! Second plot in fourth graph !
COLOR 0,255,255
POINT SYMBOL STAR
SCALE 4.0
X FUNCTION function_1
Y FUNCTION function_2
GRAPH ! Last graph !
PLOT ! Only plot in last graph !
COLOR YELLOW
BAR ON
DEPENDENT AXIS Y
X FUNCTION function_3
Y FUNCTION function_4
Following a successful completion, the finite-difference data are used to generate a second-derivative matrix. This is mass weighted and diagonalized to generate the harmonic vibrational spectrum. The second-derivative matrix (not mass-weighted) is appended to the .hessian file. Following the data for the last displacement, the flag matrix appears, followed by the lower triangle of elements of the second-derivative matrix. These data are in 5f12.7 format:
HESSIANThe data continue to H(3N,3N), where N is the number of atoms.
H(1,1)
H(2,1) H(2,2)
H(3,1) H(3,2) H(3,3)
The Discover program can output .hessian files, and the quantum programs produce and/or use files having "hessian" as part or all of their suffix.
The .hessian suffix indicates an ASCII Hessian in Discover format, and .hessianx, an ASCII Hessian in Turbomole format. Zindo, DMol and Turbomole can read both .hessian and .hessianx formats as input. Files of type .xhessian (XDR format) are no longer produced by the quantum programs (however, the quantum programs can still read them).
The following Hessian files are produced by quantum runs:
| product | calculation type | Hessian file type |
|---|---|---|
| DMol | optimization | .hessian |
| frequency | .hessian | |
| Turbomole | optimization | .hessian |
| frequency | .hessianx | |
| Zindo | optimization | .hessian |
| frequency | .hessian |
$hessian
1 1 0.6780639398 0.0000000000 0.0000000000 -0.1259825011 0.0000000000
1 2 0.0000000000 -0.2760407194 0.0000000000 -0.0947402056 -0.2760407194
1 3 0.0000000000 0.0947402056
2 1 0.0000000000 0.2160004237 0.0000000000 0.0000000000 -0.0719526695
2 2 0.0000000000 0.0000000000 -0.0720238769 0.0000000000 0.0000000000
2 3 -0.0720238769 0.0000000000
3 1 0.0000000000 0.0000000000 1.2506493403 0.0000000000 0.0000000000
3 2 -1.0175861358 -0.0926737292 0.0000000000 -0.1165316022 0.0926737292
3 3 0.0000000000 -0.1165316022
4 1 -0.1259825011 0.0000000000 0.0000000000 0.0877428007 0.0000000000
4 2 0.0000000000 0.0191198500 0.0000000000 -0.0434219786 0.0191198500
4 3 0.0000000000 0.0434219786
5 1 0.0000000000 -0.0719526695 0.0000000000 0.0000000000 0.0239681673
5 2 0.0000000000 0.0000000000 0.0239922511 0.0000000000 0.0000000000
5 3 0.0239922511 0.0000000000
6 1 0.0000000000 0.0000000000 -1.0175861358 0.0000000000 0.0000000000
6 2 1.1102928577 -0.0172186375 0.0000000000 -0.0463533610 0.0172186375
6 3 0.0000000000 -0.0463533610
7 1 -0.2760407194 0.0000000000 -0.0926737292 0.0191198500 0.0000000000
7 2 -0.0172186375 0.2759933671 0.0000000000 0.1240272754 -0.0190724977
7 3 0.0000000000 -0.0141349086
8 1 0.0000000000 -0.0720238769 0.0000000000 0.0000000000 0.0239922511
8 2 0.0000000000 0.0000000000 0.0240124468 0.0000000000 0.0000000000
8 3 0.0240191791 0.0000000000
9 1 -0.0947402056 0.0000000000 -0.1165316022 -0.0434219786 0.0000000000
9 2 -0.0463533610 0.1240272754 0.0000000000 0.1525595219 0.0141349086
9 3 0.0000000000 0.0103254413
10 1 -0.2760407194 0.0000000000 0.0926737292 0.0191198500 0.0000000000
10 2 0.0172186375 -0.0190724977 0.0000000000 0.0141349086 0.2759933671
10 3 0.0000000000 -0.1240272754
11 1 0.0000000000 -0.0720238769 0.0000000000 0.0000000000 0.0239922511
11 2 0.0000000000 0.0000000000 0.0240191791 0.0000000000 0.0000000000
11 3 0.0240124468 0.0000000000
12 1 0.0947402056 0.0000000000 -0.1165316022 0.0434219786 0.0000000000
12 2 -0.0463533610 -0.0141349086 0.0000000000 0.0103254413 -0.1240272754
12 3 0.0000000000 0.1525595219
$hessian (projected)
1 1 0.6780518841 0.0000000000 0.0000000000 -0.1259711247 0.0000000000
1 2 0.0000000000 -0.2760403797 0.0000000000 -0.0947424832 -0.2760403797
1 3 0.0000000000 0.0947424832
2 1 0.0000000000 0.2159909630 0.0000000000 0.0000000000 -0.0719448964
2 2 0.0000000000 0.0000000000 -0.0720230333 0.0000000000 0.0000000000
2 3 -0.0720230333 0.0000000000
3 1 0.0000000000 0.0000000000 1.2506493402 0.0000000000 0.0000000000
3 2 -1.0175861358 -0.0926737292 0.0000000000 -0.1165316022 0.0926737292
3 3 0.0000000000 -0.1165316022
4 1 -0.1259711247 0.0000000000 0.0000000000 0.0877339193 0.0000000000
4 2 0.0000000000 0.0191186027 0.0000000000 -0.0434183083 0.0191186027
4 3 0.0000000000 0.0434183083
5 1 0.0000000000 -0.0719448964 0.0000000000 0.0000000000 0.0239642810
5 2 0.0000000000 0.0000000000 0.0239903077 0.0000000000 0.0000000000
5 3 0.0239903077 0.0000000000
6 1 0.0000000000 0.0000000000 -1.0175861358 0.0000000000 0.0000000000
6 2 1.1102928578 -0.0172186375 0.0000000000 -0.0463533610 0.0172186375
6 3 0.0000000000 -0.0463533610
7 1 -0.2760403797 0.0000000000 -0.0926737292 0.0191186027 0.0000000000
7 2 -0.0172186375 0.2759938209 0.0000000000 0.1240265791 -0.0190720439
7 3 0.0000000000 -0.0141342123
8 1 0.0000000000 -0.0720230333 0.0000000000 0.0000000000 0.0239903077
8 2 0.0000000000 0.0000000000 0.0240163628 0.0000000000 0.0000000000
8 3 0.0240163628 0.0000000000
9 1 -0.0947424832 0.0000000000 -0.1165316022 -0.0434183083 0.0000000000
9 2 -0.0463533610 0.1240265791 0.0000000000 0.1525603398 0.0141342123
9 3 0.0000000000 0.0103246234
10 1 -0.2760403797 0.0000000000 0.0926737292 0.0191186027 0.0000000000
10 2 0.0172186375 -0.0190720439 0.0000000000 0.0141342123 0.2759938209
10 3 0.0000000000 -0.1240265791
11 1 0.0000000000 -0.0720230333 0.0000000000 0.0000000000 0.0239903077
11 2 0.0000000000 0.0000000000 0.0240163628 0.0000000000 0.0000000000
11 3 0.0240163628 0.0000000000
12 1 0.0947424832 0.0000000000 -0.1165316022 0.0434183083 0.0000000000
12 2 -0.0463533610 -0.0141342123 0.0000000000 0.0103246234 -0.1240265791
12 3 0.0000000000 0.1525603398
$end
.his is the file to which the dynamics history is periodically written during a Discover 2.9.x dynamics calculation. It is a binary file, and for a reasonable-length dynamics run it can become fairly large. It contains coordinates and other pertinent information for the system being simulated. The frequency with which this file is updated can be modified with the initialize and restart dynamics commands of the Discover 2.9.x program (see the Discover User Guide).
The .his file is written using FORTRAN unformatted I/O with the records described in Table 7. For each record, the types of the variables and the lengths of the arrays, if applicable, are given. The first frame contains extra information about the atom types, movable atoms, etc.; subsequent frames contain only the changing information--coordinates, velocities, etc.
.fhis is a formatted ASCII version of the .his file. The .fhis file is created from the .his file by the utility formhis and can be reconverted into an unformatted history file with the utility uformhis. The .fhis file is a text file that can be viewed and edited. It is also independent of a particular machine's representation of numbers and so can be transferred between dissimilar computers. The file is written using FORTRAN formatted I/O. Table 8 shows the FORTRAN format used in creating the .fhis file. A format that is enclosed in parentheses and preceded by a number indicates that the information is on more than one line, each of which has the indicated format. The number indicates the number of lines.
Special Information for the Discover 97.0/3.0.0 Program
The Discover 97.0/3.0.0 program typically sends information during dynamics runs to .arc, .out, tbl, and/or user-named files--See the Insight online help and the discussion of the output command of the Discover 2.9.7/97.0/3.0.0 User Guide for information on controlling what the Discover 97.0/3.0.0 program includes in these files and how often information is output during a dynamics run.
The layout template file contains descriptions of one or more layout templates. These templates describe the relative sizes and positions of windows in a window layout.
Layout Template File (.ltpl)
!BIOSYM layout_template 1
Layout_template:SIDE_BY_SIDE
Layout_template_type:Free_format
Layout_template_entry:
Left:0.000000
Right:50.000000
Top:0.000000
Bottom:100.000000
Layout_template_entry:
Left:50.000000
Right:100.000000
Top:0.000000
Bottom:100.000000
Layout_template:STACKED
Layout_template_type:Stacked
Stack_offset:30
Note that .mdf files are used by several Insight II products; therefore, some of the information present may be ignored by some programs or used only by certain programs.
The molecular data file has been changed minimally since the previous versions of the Discover and Insight programs. The primary change is that the potential type identifier can now be longer (up to seven characters).
Note that the order of connections listed in the .mdf file is important for atoms whose out-of-plane (oop) flag is 2 or for which chirality information is given. Therefore, these connections must not be reordered.
The .mdf file consists of one header, one end statement, and three main sections:
The sections begin and end with the character #. The order of the sections is not important, and unneeded sections can be omitted. Records within the sections begin with keyword identifiers that start with @.
In addition, comment records, beginning with !, are allowed.
The overall structure of an .mdf file is shown in Table 9. Descriptions and examples of each major part follow.
Header Record
The first record of a molecular data file must be:
!BIOSYM molecular_data #The ! must be the first character in the file. The Discover program interprets this line as indicating an ASCII file containing molecular data records as outlined in this section. The string molecular_data indicates that the contents of the file are those of an .mdf file; the # is replaced by an actual number, which identifies the file format for the Discover program. The number 4, for example, indicates that the file format is as specified here for the Discover program, versions 2.9.5/3.2 and later.
#topologyNext, the column headings are defined. The molecule name and atomic data follow.
Column records have the following syntax:
@column # type specifier
where @column is a keyword identifying the record, # is the number of a column containing a certain type of atomic data, and specifier (for example, the name of a forcefield) further defines the type, when necessary.
The types of atomic data are shown in Table 9. Column headings must all be listed, in the order given.
@column 1 element
@column 2 atom_type cvff
...
@molecule name type
where @molecule is an identifying keyword, name is a molecule name for identification purposes, and type is the optional type of molecule for classification purposes. If type is present, all molecules of the same type must be topologically identical.
Examples:
@molecule crambin
@molecule wat4 water
@molecule h2o5 water
@molecule benz1 c6h6
Atom records consist of the fields shown in Table 10. The values allowed for flag settings are also shown.
The syntax for atom records consists of one record for each of the first 11 data types listed in the table, followed on the same line by the connectivity records, which consist of several records.
12345678901234567890123456789012345678901234567890123456789012345678901234567890
ACE_1:CA C c3 meA 0 0 -0.3000 1 0 8 1.0000 0.0000 4 connectivity record(The underlined numbers are used merely to indicate the column numbers--they are not part of the file.)
N-M_3:N N n nme 0 0 -0.5000 1 1 8 1.0000 0.0000 3 connectivity record
resname_resnumber:atom%cellxyz#symop/bondorder,wedgebond
The number of connectivity records equals the number of atoms (including ghost atoms) that that atom is connected to. The meaning and default values of each of these is shown in Table 11. Except for atom and /bondorder, all other portions of a record may be omitted if the default values are satisfactory.
The order in which the atoms are listed in the connectivity record should correspond to the chirality flag. The priority ordering used for determining chirality and prochirality is reflected by the listing order from lowest to highest priorities.
LEU_6:N%000#1/2.0 N/2.0
LEU_6:N%+0-1+0#1/1.0 N%0-10/1.0
LEU_6:N%000#5/2.0 N#5/2.0
ALA_7:N%000#1/1.0 ALA_7:N/1.0
eth_1:C1 C c a 0 0 -0.1200 1 0 1 4 C2 C3%001This example is for an isolated 3-atom helix containing all single bonds. Atom C1 is connected to atom C2 and to a helical image of atom C3 so that C1 lies between C3 and the image of C3. Likewise, C3 is bonded to atom C2 and to the helical image of C1. The locations of the image atoms are generated by means of helix information stored in the .car or .arc files.
eth_1:C2 C c a 0 0 -0.1200 1 0 1 4 C1 C2
eth_1:C3 C c b 0 0 -0.1200 1 0 1 4 C2 C1%00-1
#symmetryThis is followed by records describing the periodicity, the symmetry group associated with the periodicity, explicit matrix representations of the operators of the group, and whether helical symmetry is present. Some of these records are optional.
The syntax of the periodicity record is:
@periodicity type axes
where @periodicity is the keyword identifier, type is the number of dimensions in which translational periodicity occurs, and axes is a description of how the periodic vectors relate to the Cartesian coordinate system. The type can have the values shown in Table 12.
| value of periodicity type | definition |
|---|---|
| 0 | no periodicity (default) |
| 2 | 2D periodicity |
| 3 | 3D periodicity |
@periodicity 3 xyz @periodicity 2 xy @periodicity 3The last entry implies that the default axes specification is used.
The syntax of the group record is:
@group name or @group matrix #
where @group is the keyword identifier, name is the symmetry group name, matrix is a keyword indicating that matrix representations of the operators follow, and # is the number of matrices.
The symmetry group name is that associated with the periodicity type. For example, if the periodicity type is 3, then the group name is the name of a space group (see the Discover 2.9.x/97.0/3.0.0 User Guide). If the periodicity type is 2, then the group name is the name of a plane group.
Likewise, the matrix representations that follow are those for the operators associated with the given periodicity type.
Examples:
@group (P21 21 2)
@group matrix 4
The syntax of a matrix record is:
@matrix # name
where @matrix is the keyword identifier, # is the number of the operator and runs from 1 continuously to the number of operators in the group, name is an optional name for the operator. The single letters on the next four lines are the individual floating-point elements of the 4 X 4 representation matrix of the operator.
All matrices, including the identity operator, must be specified.
Example:
! Space group 5 (C 2)
@matrix 1
1. 0. 0. 0.
0. 1. 0. 0.
0. 0. 1. 0.
0. 0. 0. 1.
@matrix 2
-1. 0. 0. 0.
0. 1. 0. 0.
0. 0. -1. 0.
0. 0. 0. 1.
@matrix 3
1. 0. 0. 0.
0. 1. 0. 0.
0. 0. 1. 0.
0.5 0.5 0. 1.
@matrix 4
-1. 0. 0. 0.
0. 1. 0. 0.
0. 0. -1. 0.
0.5 0.5 0. 1.
The syntax of the helix record is:
@helix
where @helix is the keyword identifier.
A helix record can be present only when the periodicity type is 0 or 2. Currently, the presence of this record means that all molecules in the system have helical symmetry.
Helix information is currently used only by the Polymer programs. The Discover program ignores the helix record, since it does not currently support infinite helices. The Insight program uses the helix record to display helical systems.
Examples:
#symmetry
@periodicity 2 xy
@helix #symmetry
@helix
#atomsetEach atomset record is introduced by a line having the following syntax:
@degree type name [other]
where @degree is an integer or synonymous word indicating how many subsequent atoms make up a single entry. For example, a list of bonds or distances have a degree of 2, and a list of dihedral angles has a degree of 4. A general list, with no association of atoms, has a degree of 1. The synonyms list, pair, triplet, and quartet are used for degrees 1 through 4, respectively.
The type field specifies a general type for the set of atoms and is used in determining how the set is to be used (Table 14).
The name field is the identifying name given to the set of atoms. Depending on the type of set, this can be the name of a torsion, pseudoatom, backbone, or general subset.
poly1:eth1:c1 c2 eth2:c1 c2 poly2:eth1:c1 c2is equivalent to the following explicit list:
poly1:eth1:c1 poly1:eth1:c2 poly1:eth2:c1 poly1:eth2:c2
poly2:eth1:c1 poly2:eth1:c2
@list backbone name
where name is used to identify the backbone.
The atom list contains zero or more atoms and can continue for more than one line without any explicit continuation characters--it is considered finished at the next @ or # symbol. The specification for the first atom is in the standard Insight format. If the molecule or residue portions of the specifications for the other atoms are missing, they default to the previous appropriate value used in the list. Wildcards are allowed.
Example of defining backbone atoms:
@list backbone 1
poly1:eth_1:C1 C2
poly1:sty_2:C1 C2
@list subset name
where name is used to identify the subset. The atom list is the same as for defining backbone atoms.
Example of defining subset atoms:
@list subset eth1
poly1:eth1:c1 c2 h1 h2 h3 h4
@list pseudoatom name A
where name is the name for the pseudoatom in the form of a simple atom specification, and A (arithmetic average) indicates the method of calculating pseudoatom coordinates. The atom list is the same as for defining backbone atoms.
Examples of defining pseudoatoms:
@list pseudoatom xmol:xres:x A
*:*:* @list pseudoatom water:xres:cm A
water:*:* @list pseudoatom poly1:sty_2:XPHE A
poly1:sty_2:C3 C4 C5 C6 C7 C8
H3 H4 H5 H6 H7 H8 @list pseudoatom poly1:xres:x A
poly1:eth_1:C* sty_2:C1,C2
@quartet torsion name
where name is the name of the torsion being defined and must include the molecule and residue names in the standard Insight format. Wildcards are allowed. The molecule and residue names can be omitted from the atom names, in which case they are assumed to be the same as in the torsion name. Relative residue numbers denoted by a signed integer may be used (e.g., -1:C or +1:N). Full molecule and residue names may be given, but must also be used in the torsion name.
Examples of defining torsions:
@quartet torsion *:*:phi
-1:C N CA C @quartet torsion *:VAL_*:chi1
N CA CB CG1 @quartet torsion crn:tor:tors
crn:1:C 2:N 2:CA 2:HA
!BIOSYM molecular_data 4
!
!DATE: Fri Sep 27 13:50:15 1993 INSIGHT generated molecular data file
!
#topology
!
@column 1 element
@column 2 atom_type cvff
@column 3 charge_group cvff
@column 4 isotope
@column 5 formal_charge
@column 6 charge cvff
@column 7 switching_atom cvff
@column 8 oop_flag cvff
@column 9 chirality_flag
@column 10 occupancy
@column 11 xray_temp_factor
@column 12 connections
!
@molecule ACEALANM
!
ACE_1:CA C c3 meA 0 0 -0.3000 1 0 8 1.0000 0.0000 HA1 HA2 HA3 C
ACE_1:HA1 H h meA 0 0 0.1000 0 0 8 1.0000 0.0000 CA
ACE_1:HA2 H h meA 0 0 0.1000 0 0 8 1.0000 0.0000 CA
ACE_1:HA3 H h meA 0 0 0.1000 0 0 8 1.0000 0.0000 CA
ACE_1:C C c' pepC 0 0 0.3800 1 1 8 1.0000 0.0000 CA O/2.0 ALA_2:N
ACE_1:O O o' pepC 0 0 -0.3800 0 0 8 1.0000 0.0000 C/2.0
ALA_2:N N n pepN 0 0 -0.5000 1 1 8 1.0000 0.0000 ACE_1:C CA HN
ALA_2:CA C ca pepN 0 0 0.1200 0 0 8 1.0000 0.0000 N HA C CB
ALA_2:HN H hn pepN 0 0 0.2800 0 0 8 1.0000 0.0000 N
ALA_2:HA H h pepN 0 0 0.1000 0 0 8 1.0000 0.0000 CA
ALA_2:C C c' pepC 0 0 0.3800 1 1 8 1.0000 0.0000 CA O/2.0 N-M_3:N
ALA_2:O O o' pepC 0 0 -0.3800 0 0 8 1.0000 0.0000 C/2.0
ALA_2:CB C c3 meB 0 0 -0.3000 1 0 8 1.0000 0.0000 CA HB1 HB2 HB3
ALA_2:HB1 H h meB 0 0 0.1000 0 0 8 1.0000 0.0000 CB
ALA_2:HB2 H h meB 0 0 0.1000 0 0 8 1.0000 0.0000 CB
ALA_2:HB3 H h meB 0 0 0.1000 0 0 8 1.0000 0.0000 CB
N-M_3:N N n nme 0 0 -0.5000 1 1 8 1.0000 0.0000 ALA_2:C CA HN
N-M_3:CA C c3 nme 0 0 -0.0800 0 0 8 1.0000 0.0000 N HA1 HA2 HA3
N-M_3:HN H hn nme 0 0 0.2800 0 0 8 1.0000 0.0000 N
N-M_3:HA1 H h nme 0 0 0.1000 0 0 8 1.0000 0.0000 CA
N-M_3:HA2 H h nme 0 0 0.1000 0 0 8 1.0000 0.0000 CA
N-M_3:HA3 H h nme 0 0 0.1000 0 0 8 1.0000 0.0000 CA
!
#atomset
!
@quartet torsion *:ALA_2:omeg
CA C *:N *:CA
@quartet torsion *:ALA_2:phi
*:C N CA C
@quartet torsion *:ALA_2:chi1
N CA CB HB1
!BIOSYM molecular_data 4
!DATE: Thu Jun 11 15:24:13 1993 INSIGHT generated molecular data file
!
#topology
!
@column 1 element
@column 2 atom_type cvff
@column 3 charge_group cvff
@column 4 isotope
@column 5 formal_charge
@column 6 charge cvff
@column 7 switching_atom cvff
@column 8 oop_flag cvff
@column 9 chirality_flag
@column 10 n_connections
@column 11 connectivity
!
@molecule WTR1 water
WTR_1:O1 O o* WTR 16 0 -0.8200 1 0 0 2 H1 H2
WTR_1:H1 H h* WTR 2 0 0.4100 0 0 0 1 O1
WTR_1:H2 H h* WTR 2 0 0.4100 0 0 0 1 O1
!
@molecule SF6 sulfur_hexafluoride
sf6_1:S S s a 0 1+ 1.5000 1 0 0 6 F1 F1#2 F1#3 F1#4 F2 F3
sf6_1:F1 F f a 0 1/6- -0.2500 0 0 0 1 S
sf6_1:F2 F f a 0 1/6- -0.2500 0 0 0 1 S
sf6_1:F3 F f a 0 1/6- -0.2500 0 0 0 1 S
!
@molecule poly1 ethylene-styrene
eth_1:C1 C c a 0 0 -0.1200 1 0 1 4 H1 H2 C2 sty_2:C2%010
eth_1:H1 H h a 0 0 0.0600 0 0 0 1 C1
eth_1:H2 H h a 0 0 0.0600 0 0 0 1 C1
eth_1:C2 C c b 0 0 -0.1200 1 0 1 4 H3 H4 C1 sty_2:C1
eth_1:H3 H h b 0 0 0.0600 0 0 0 1 C2
eth_1:H4 H h b 0 0 0.0600 0 0 0 1 C2
sty_2:C1 C c me1 0 0 -0.0600 1 0 4 4 H1 C2 eth_1:C2 sty_2:C3
sty_2:H1 H h me1 0 0 0.0600 0 0 0 1 C1
sty_2:C2 C c me2 0 0 -0.1200 1 0 1 4 H2 H3 C1 eth_1:C1%0-10
sty_2:H2 H h me2 0 0 0.0600 0 0 0 1 C2
sty_2:H3 H h me2 0 0 0.0600 0 0 0 1 C2
sty_2:C3 C cp ph1 0 0 0.0000 1 1 0 3 C1 C4/1.5 C8/1.5
sty_2:C4 C cp ph1 0 0 -0.1000 0 1 0 3 C3/1.5 C5/1.5 H4
sty_2:H4 H h ph1 0 0 0.1000 0 0 0 1 C4
sty_2:C8 C cp ph1 0 0 -0.1000 0 1 0 3 C3/1.5 C7/1.5 H8
sty_2:H8 H h ph1 0 0 0.1000 0 0 0 1 C8
sty_2:C5 C cp ph2 0 0 -0.1000 0 1 0 3 C4/1.5 C6/1.5 H5
sty_2:H5 H h ph2 0 0 0.1000 0 0 0 1 C5
sty_2:C6 C cp ph2 0 0 -0.1000 1 1 0 3 C5/1.5 C7/1.5 H6
sty_2:H6 H h ph2 0 0 0.1000 0 0 0 1 C6
sty_2:C7 C cp ph2 0 0 -0.1000 0 1 0 3 C6/1.5 C8/1.5 H7
sty_2:H7 H h ph2 0 0 0.1000 0 0 0 1 C7
!
#symmetry
@periodicity 3 xyz
@group matrix 4
@matrix 1 identity
1.000 0.000 0.000 0.000
0.000 1.000 0.000 0.000
0.000 0.000 1.000 0.000
0.000 0.000 0.000 1.000
@matrix 2 C4_1
0.000 1.000 0.000 0.000
1.000 0.000 0.000 0.000
0.000 0.000 1.000 0.000
0.000 0.000 0.000 1.000
@matrix 3 C4_2
-1.000 0.000 0.000 0.000
0.000 1.000 0.000 0.000
0.000 0.000 1.000 0.000
0.000 0.000 0.000 1.000
@matrix 4 C4_3
0.000 -1.000 0.000 0.000
-1.000 0.000 0.000 0.000
0.000 0.000 1.000 0.000
0.000 0.000 0.000 1.000
!BIOSYM molecular_data 4
!DATE: Thu Jun 11 17:44:53 1993 INSIGHT generated molecular data file
#topology
@column 1 element
@column 2 atom_type cvff
@column 3 charge_group cvff
@column 4 isotope
@column 5 formal_charge
@column 6 charge cvff
@column 7 switching_atom cvff
@column 8 oop_flag cvff
@column 9 chirality_flag
@column 10 occupancy
@column 11 xray_temp_factor
@column 12 connections
@molecule TEST_6_11_HLX1
ETHE_1:C1 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H11 H12 C2 C2%001
ETHE_1:H11 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:H12 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:C2 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H21 H22 C1 C1%00-1
ETHE_1:H21 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
ETHE_1:H22 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
#symmetry
@helix
!
#atomset
@list backbone TEST_6_11_HLX1
TEST_6_11_HLX1:ETHE_1:C1 C2
!BIOSYM molecular_data 4
!DATE: Thu Jun 11 17:42:58 1993 INSIGHT generated molecular data file
#topology
@column 1 element
@column 2 atom_type cvff
@column 3 charge_group cvff
@column 4 isotope
@column 5 formal_charge
@column 6 charge cvff
@column 7 switching_atom cvff
@column 8 oop_flag cvff
@column 9 chirality_flag
@column 10 occupancy
@column 11 xray_temp_factor
@column 12 connections
@assembly NEW_CELL
@molecule TEST_6_11_HLX
ETHE_1:C1 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H11 H12 C2 C2%001
ETHE_1:H11 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:H12 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:C2 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H21 H22 C1 C1%00-1
ETHE_1:H21 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
ETHE_1:H22 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
@molecule TEST_6_11_HLX01
ETHE_1:C1 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H11 H12 C2 C2%001
ETHE_1:H11 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:H12 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:C2 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H21 H22 C1 C1%00-1
ETHE_1:H21 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
ETHE_1:H22 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
@molecule TEST_6_11_HLX0101
ETHE_1:C1 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H11 H12 C2 C2%001
ETHE_1:H11 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:H12 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:C2 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H21 H22 C1 C1%00-1
ETHE_1:H21 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
ETHE_1:H22 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
@molecule TEST_6_11_HLX010101
ETHE_1:C1 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H11 H12 C2 C2%001
ETHE_1:H11 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:H12 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C1
ETHE_1:C2 C c2 0 0 0 1.0000 0 1 8 0.0000 0.0000 H21 H22 C1 C1%00-1
ETHE_1:H21 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
ETHE_1:H22 H h 0 0 0 0.0000 0 0 8 0.0000 0.0000 C2
#symmetry
@periodicity 2 xy
@group (P1)
@helix
!
#atomset
@list backbone TEST_6_11_HLX
TEST_6_11_HLX:ETHE_1:C1 C2
@list backbone TEST_6_11_HLX01
TEST_6_11_HLX01:ETHE_1:C1 C2
@list backbone TEST_6_11_HLX0101
TEST_6_11_HLX0101:ETHE_1:C1 C2
@list backbone TEST_6_11_HLX010101
TEST_6_11_HLX010101:ETHE_1:C1 C2
The file reader from Insight releases earlier than Insight II 97.0 ignore the new records and the changes to the existing record types do not impact the reader.
The Insight II 97.0 file reader handles the new parts of the PDB file format in the following ways.
The .pdbx file is essentailly similar to the Brookhaven Protein Data Bank (PDB) format, with the following differences:
!BIOSYM nmr_peak_intensities 2
!
#mixing_times
2.000000E-02 4.000000E-02 8.000000E-02 1.200000E-01
!
#peak_intensities
!Peak W2_Pos W1_Pos LineWdth2 LineWdth1 Intensities
!
120 5.315 2.091 8.575 17.070 4.0410E+05 2.3580E+05 6.4820E+05 4.1640E+05
121 5.256 3.133 8.575 17.790 1.9220E+06 2.6670E+06 3.3220E+06 3.8150E+06
122 5.123 1.009 8.575 17.910 8.4710E+06 1.1300E+07 2.3800E+07 3.0970E+07
123 4.324 2.344 10.020 17.600 5.8450E+05 2.0510E+06 4.7590E+06 7.6090E+06
124 4.081 4.400 9.328 16.770 3.3020E+05 -1.3270E+05 2.8710E+05 1.2310E+06
125 4.006 1.212 8.575 16.890 -6.1340E+05 7.5890E+05 1.3390E+06 2.7490E+06
126 3.336 1.003 9.005 17.560 -8.9400E+04 -7.3900E+05 2.0200E+06 2.5500E+06
127 4.081 8.570 8.575 16.890 4.7400E+06 1.2700E+07 2.3900E+07 3.1900E+07
128 5.235 1.789 9.320 25.200 4.6700E+05 7.3400E+05 9.7000E+05 2.0400E+06
129 2.323 0.763 8.575 17.070 2.4600E+06 4.4700E+06 6.6900E+06 9.0400E+06
130 3.456 1.234 8.575 17.790 -1.1000E+05 -4.1000E+04 3.1600E+05 1.3900E+06
131 5.289 2.569 8.575 17.910 3.16000E+05 6.6000E+06 1.5400E+07 2.1000E+07
132 1.312 1.132 10.020 17.600 5.3800E+06 8.3500E+06 1.1390E+07 1.6870E+07
133 1.789 1.766 9.328 16.770 3.3800E+06 8.3500E+06 1.1390E+07 1.6870E+07
134 3.232 2.737 8.575 16.890 9.4200E+05 2.6000E+06 8.6100E+06 1.2400E+07
!BIOSYM pseudo_atom_library 1
!
! CVFF
!
#plb_entry
ALAN Alanine, positive N-terminus
HNX
HN1 HN2 HN3
Type NH3
Not_Prochiral
!
HBX
HB1 HB2 HB3
Type CH3
Not_Prochiral
!
!
#plb_entry
ALA Alanine, polypeptide residue
HBX
HB1 HB2 HB3
Type CH3
Not_Prochiral
!
!
.
.
.
#plb_entry
LEUN Leucine, positive N-terminus
HNX
HN1 HN2 HN3
Type NH3
Not_Prochiral
!
HBX
HB1 HB2
Type CH2
Not_Prochiral
!
HD1X
HD11 HD12 HD13
Type CH3
Prochiral Center: CG Reference: CD1
!
HD2X
HD21 HD22 HD23
Type CH3
Prochiral Center: CG Reference: CD2
!
HDX
HD11 HD12 HD13 HD21 HD22 HD23
Type 2CH3
Not_Prochiral
!
!
.
.
.
| Section | Example | Description |
|---|---|---|
| Header | !BIOSYM nmr_chemical_shifts 1 | |
| chemical shifts | <atom_spec> <float1> <float2> <float3> | Chemical shift information |
| Variable | Description |
|---|---|
|
<atom_spec>
| reference to a hydrogen or pseudoatom |
|
<float1>
| chemical shift in ppm |
|
<float2>
| T1 leakage rate |
|
<float3>
| Line width in Hz |
1:ASN_1:HBR 1.1500 0.0000 0.0000
!BIOSYM nmr_chemical_shifts 1
#chemical_shifts
! Atom Spec PPM T1 Leak Line Width
!
1:SERN_1:HA 4.390 1.000 20.000
1:SERN_1:HB* 4.080 1.000 20.000
1:SERN_1:HG 1.100 1.000 20.000
1:ASN_2:HN 8.570 1.000 20.000
1:ASN_2:HA 4.550 1.000 20.000
1:ASN_2:HB2 2.740 1.000 20.000
1:ASN_2:HB1 3.230 1.000 20.000
1:ASN_2:HD2* 6.710 1.000 20.000
1:PHE_3:HN 9.300 1.000 20.000
1:PHE_3:HA 4.040 1.000 20.000
1:PHE_3:HB* 3.570 1.000 20.000
1:PHE_3:HD* 7.230 1.000 20.000
1:PHE_3:HE* 7.390 1.000 20.000
1:PHE_3:HZ 7.270 1.000 20.000
...
1:AR+C_7:HD* 2.405 1.000 20.000
The pro_angle.dat file contains a protein bond angle table consisting of residue- specific bond angles and standard deviations. This information was derived from
File rules:
C N CA * GLY GLY 120.6 1.7describes a C-N-CA bond angle (where the carbonyl atom may be in any residue while the nitrogen and alpha carbon atoms are in a glycine) as having a mean bond angle of 120.6° with a standard deviation of 1.7°.
! Created Sept 7 1994.
! Residue Specific bond Angles and standard deviations
! Information derived from
! R.A.Engh and R.Huber, Acta. Cryst., A47 292-300 (1991),
! File rules
! 1) Later lines take precedence over earlier ones with
! atoms of same names.
! 2) Atom name in column 1 is associated with residue name
! in column 4, 2 with 5 etc.
! 3) Only wildcarding allowed is a single * character
! this implies a match with any residue name
! 4) A zero entry implies this specific bond not present
! in the data base and will not be checked.
C N CA * * * 121.7 1.8
C N CA * GLY GLY 120.6 1.7
C N CA * PRO PRO 122.6 5.0
CA C N * * * 116.2 2.0
CA C N GLY GLY * 116.4 2.1
CA C N * * PRO 116.9 1.5
CA C N GLY GLY PRO 118.2 2.1
CA C O * * * 120.8 1.7
CA C O GLY GLY GLY 120.8 2.1
CB CA C * * * 110.1 1.9
CB CA C ALA ALA ALA 110.5 1.5
CB CA C ILE ILE ILE 109.1 2.2
CB CA C THR THR THR 109.1 2.2
CB CA C VAL VAL VAL 109.1 2.2
N CA C * * * 111.2 2.8
N CA C * GLY GLY 112.5 2.9
N CA C PRO * * 111.8 2.5
N CA C PRO GLY GLY 0.0 0.0
N CA CB * * * 110.5 1.7
N CA CB ILE ILE ILE 111.5 1.7
N CA CB THR THR THR 111.5 1.7
N CA CB VAL VAL VAL 111.5 1.7
N CA CB ALA ALA ALA 110.4 1.5
N CA CB PRO PRO PRO 103.0 1.1
O C N * * * 123.0 1.6
O C N * * PRO 122.0 1.4
The pro_bond.dat file contains a protein bond length table consisting of residue-specific bond lengths and standard deviations. This information was derived from
File rules:
N CA GLY 1.451 0.016states that the bond between a Glycine nitrogen and an alpha carbon has a mean value of 1.451 Å, with a standard deviation of 0.016 Å.
Much of the following information is taken straight from the files themselves and serves to explain the syntax and meaning of the values.
The pro_bond.dat file contains a protein bond length table consisting of residue-specific bond lengths and standard deviations. This information was derived from
File rules:
N CA GLY 1.451 0.016states that the bond between a Glycine nitrogen and an alpha carbon has a mean value of 1.451 Å, with a standard deviation of 0.016 Å.
| Variable | Description |
|---|---|
|
<atom1>
| Valid atom name |
|
<atom2>
| Valid atom name |
|
<residue>
| Valid residue name |
|
<mean>
| Bond length mean value in Å |
|
<std_devn>
| Bond length standard deviation in Å |
! Created Sept 7 1994.
! Residue Specific bond lengths and standard deviations
! Information derived from
! R.A.Engh and R.Huber, Acta. Cryst., A47 292-300 (1991),
! File rules
! 1) Later lines take precedence over earlier ones with
! atoms of same names.
! 2) Atom name in first column is associated with residue name
! in third column.
! 3) Only wildcarding allowed is a single * character
! this implies a match with any residue name
CA C * 1.525 0.021
CA C GLY 1.516 0.018
C O * 1.231 0.020
CB CA * 1.530 0.020
CB CA ALA 1.521 0.033
CB CA ILE 1.540 0.027
CB CA THR 1.540 0.027
CB CA VAL 1.540 0.027
N CA * 1.458 0.019
N CA PRO 1.466 0.015
N CA GLY 1.451 0.016
N C * 1.329 0.014
N C PRO 1.341 0.016
SG SG CYS 2.000 0.100
!BIOSYM project 1
Mon Nov 4 11:04:14 1991
RMA Run: test Molecule=CRAM7AVG NMR_project=test.
Test of average structure.
Project files written:
cram7avg.ppm cram7avg.pks cram7avg.asn cram7avg.rstrnt
RMA files written:
test.rmainp test.mdh test.shift test.rma_temp test.rstrnt_temp
Updated RMA files are test_01.rma and test_01.rstrnt
!BIOSYM project 1
Mon Nov 4 11:58:29 1991
RMA Run: test Molecule=CRAM7AVG NMR_project=test.
Test of average structure.
Project files written:
cram7avg.ppm cram7avg.pks cram7avg.asn cram7avg.rstrnt
RMA files written:
test.rmainp test.mdh test.shift test.rma_temp test.rstrnt_temp
Updated RMA files are test_02.rma and test_02.rstrnt
The pro_misc.dat file contains a table of miscellaneous protein properties. This information was derived from J. Thornton and co workers (J. Appl. Cryst., 26, 283-291, 1993), R. A. Engh and R. Huber (Acta. Cryst., A47, 292-300, 1991), and from M. Macarthur (private communication).
File rules:
OMEGA 180.0 5.8describes the omega torsion angle as having a mean value of 180.0 °, with a standard deviation of 5.8 °.
Much of the following information is taken straight from the files themselves and serves to explain the syntax and meaning of the values.
The pro_misc.dat file contains a table of miscellaneous protein properties. This information was derived from J. Thornton and co workers (J. Appl. Cryst., 26, 283-291, 1993), R. A. Engh and R. Huber (Acta. Cryst., A47, 292-300, 1991), and from M. Macarthur (private communication).
File rules:
OMEGA 180.0 5.8describes the omega torsion angle as having a mean value of 180.0 °, with a standard deviation of 5.8 °.
| Section | Example | Description |
|---|---|---|
| Comment | !File rules | ! character implies comment |
| Data | <keyword> <mean> <std_devn> | Property name, mean value, and standard deviation |
| Variable | Description |
|---|---|
|
<keyword>
| Name of per-residue property |
|
<mean>
| Property mean value |
|
<std_devn>
| Property standard deviation |
! Created Sept 15 1994.
! Contains information on miscellaneous protein properties.
! Information derived from
! J. Thornton and co workers J. Appl. Cryst. vol 26, 283-291 (1993)
! R.A.Engh and R.Huber, Acta. Cryst., A47 292-300 (1991),
! M.Macarthur private communication.
! File rules
! 1) First word on line is property keyword
! second word is property mean value
! third word is property standard deviation
! 2) Values of mean and standard deviation can be modified by user
! names of keywords cannot be changed
! 3) Unrecognised keywords ignored.
!
! Kabsch+Sander H-Bond energies in Kcal
CHI_1_RANGE_1 64.1 15.7
CHI_1_RANGE_2 183.6 16.8
CHI_1_RANGE_3 -66.7 15.0
CHI_2_RANGE_1 68.7 21.3
CHI_2_RANGE_2 177.5 19.4
CHI_2_RANGE_3 -71.8 21.1
PROLINE_PHI -65.4 11.2
HELIX_PHI -65.3 11.9
HELIX_PSI -39.4 11.3
CHI_3_SS_RANGE_1 96.8 14.8
CHI_3_SS_RANGE_2 -85.8 10.7
OMEGA 180.0 5.8
E_H_BOND_KS -2.02 0.75
CA_VIRTUAL_TORSION 33.9 3.5
WRITe STRUcturestement in X-PLOR and is suitable for input using the
STRUcturestatement in X-PLOR. The contents of this file consist of atmo names, types, charges and masses; residue names and segment names; and a list of bond terms, angle terms, dihedral terms, improper terms, explicit hydrogen-bonding terms, explicit nonbonded exclusions, and nonbonded group partitions. It does not contain atomic coordinates,parameters, constraints, restraints, or any other information that is specific to effective energy terms, such as diffraction data.
Refer to Chapter 3 of X-PLOR, Version 3.1, A System for X-ray Crystallography and NMR (Axel Brunger, Yale Univ. Press, 1992) for the description and usage of this file format.
Three standard residue libraries are provided:
Note: A complete description of the residue library is given below. However, only the potential function atom type, partial charge, charge group, and named torsion fields are used by Insight II. All other numeric fields may be set to 0 for use with Insight II. Insight II uses the geometry, topology, and bond order information found in the fragment libraries for all building functions.
The residue library provides connectivity information by specifying the parent of each and every atom found in the molecule. For each atom, the parent of the atom is uniquely specified, the bond order is given, and if the atom is involved in a ring closure then the ring closure atom is also specified. If the parent of a given atom is found in the current residue then the name of that parent atom is given. If the parent is in the preceding residue then the bond order to the parent atom is followed by an asterisk, *, to indicate that the parent is located in the previous residue.
The three geometrical parameters that are provided for each atom are:
The next two fields contain two flags:
The next two items specified for each atom in the residue library are:
The final two fields are used to identify the charge group to which the atom belongs and whether or not this atom is the switching atom (i.e., the atom used to decide whether this charge group is within the cutoff distance for nonbond calculations). A portion of Insight's residue library is given as an illustration in Portion of Insight's Residue Library. (Note that the first two lines shown below are used merely to label the column numbers; they are not part of the library.) The residue libraries are found in the directory pointed to by the environment variable $BIOSYM_LIBRARY.
Portion of Insight's Residue Library
(This excerpt does not include the beginning of the file.)
1 2 3 4 5 6 7 8
12345678901234567890123456789012345678901234567890123456789012345678901234567890
SD CG 1.0 1.740 111.000 195.000 chi2 1 0 s 0.1200 csc 1
CE SD 1.0 1.670 101.000 194.000 chi3 1 0 c3 -0.3200 csc 0
HE1 CE 1.0 1.080 110.000 300.000 0 0 h 0.1000 csc 0
HE2 CE 1.0 1.080 110.000 180.000 chi4 1 0 h 0.1000 csc 0
HE3 CE 1.0 1.080 110.000 60.000 0 0 h 0.1000 csc 0
PHE 20 Phenylalanine, polypeptide residue
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
CA N 1.0 1.436 123.100 180.000 omeg 0 0 ca 0.1200 pepN 0
HN N 1.0 1.080 123.000 0.000 0 0 hn 0.2800 pepN 0
HA CA 1.0 1.080 110.000 300.000 0 0 h 0.1000 pepN 0
C CA 1.0 1.509 109.600 180.000 phi 0 1 c' 0.3800 pepC 1
O C 2.0 1.263 118.100 0.000 0 0 o' -0.3800 pepC 0
CB CA 1.0 1.554 111.600 60.000 0 0 c2 -0.2000 meB 1
HB1 CB 1.0 1.080 110.000 63.000 0 0 h 0.1000 meB 0
HB2 CB 1.0 1.080 110.000 183.000 0 0 h 0.1000 meB 0
CG CB 1.0 1.472 113.700 303.000 chi1 1 1 cp 0.0000 arG 1
CD1 CG 1.5 1.376 123.100 87.400 chi2 1 1 cp -0.1000 arD1 1
HD1 CD1 1.0 1.080 120.000 0.000 0 0 h 0.1000 arD1 0
CE1 CD1 1.5 1.368 122.600 180.000 0 1 cp -0.1000 arE1 1
HE1 CE1 1.0 1.080 120.000 180.000 0 0 h 0.1000 arE1 0
CZ CE1 1.5 1.388 118.900 0.000 0 1 cp -0.1000 arZ 1
HZ CZ 1.0 1.080 120.000 180.000 0 0 h 0.1000 arZ 0
CE2 CZ 1.5 1.380 120.600 0.000 0 1 cp -0.1000 arE2 1
HE2 CE2 1.0 1.080 120.000 180.000 0 0 h 0.1000 arE2 0
CD2 CE2 1.5 CG 1.5 1.376 118.000 0.000 0 1 cp -0.1000 arD2 1
HD2 CD2 1.0 1.080 120.000 180.000 0 0 h 0.1000 arD2 0
PHEn 21 Phenylalanine, neutral N-terminus
N N 1.0 0.000 0.000 0.000 0 0 n2 -0.5000 pepN 1
CA N 1.0 1.436 0.000 0.000 0 0 ca 0.1200 pepN 0
HN1 N 1.0 1.080 123.000 0.000 0 0 hn 0.1400 pepN 0
HN2 N 1.0 1.080 123.000 180.000 0 0 hn 0.1400 pepN 0
HA CA 1.0 1.080 110.000 120.000 0 0 h 0.1000 pepN 0
C CA 1.0 1.509 109.600 0.000 0 1 c' 0.3800 pepC 1
SD CG 1.0 1.740 111.000 195.000 chi2 1 0 s 0.1200 csc 1
CE SD 1.0 1.670 101.000 194.000 chi3 1 0 c3 -0.3200 csc 0
HE1 CE 1.0 1.080 110.000 300.000 0 0 h 0.1000 csc 0
HE2 CE 1.0 1.080 110.000 180.000 chi4 1 0 h 0.1000 csc 0
HE3 CE 1.0 1.080 110.000 60.000 0 0 h 0.1000 csc 0
PHE 20 Phenylalanine, polypeptide residue
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
CA N 1.0 1.436 123.100 180.000 omeg 0 0 ca 0.1200 pepN 0
HN N 1.0 1.080 123.000 0.000 0 0 hn 0.2800 pepN 0
HA CA 1.0 1.080 110.000 300.000 0 0 h 0.1000 pepN 0
C CA 1.0 1.509 109.600 180.000 phi 0 1 c' 0.3800 pepC 1
O C 2.0 1.263 118.100 0.000 0 0 o' -0.3800 pepC 0
CB CA 1.0 1.554 111.600 60.000 0 0 c2 -0.2000 meB 1
HB1 CB 1.0 1.080 110.000 63.000 0 0 h 0.1000 meB 0
HB2 CB 1.0 1.080 110.000 183.000 0 0 h 0.1000 meB 0
CG CB 1.0 1.472 113.700 303.000 chi1 1 1 cp 0.0000 arG 1
CD1 CG 1.5 1.376 123.100 87.400 chi2 1 1 cp -0.1000 arD1 1
HD1 CD1 1.0 1.080 120.000 0.000 0 0 h 0.1000 arD1 0
CE1 CD1 1.5 1.368 122.600 180.000 0 1 cp -0.1000 arE1 1
HE1 CE1 1.0 1.080 120.000 180.000 0 0 h 0.1000 arE1 0
CZ CE1 1.5 1.388 118.900 0.000 0 1 cp -0.1000 arZ 1
HZ CZ 1.0 1.080 120.000 180.000 0 0 h 0.1000 arZ 0
CE2 CZ 1.5 1.380 120.600 0.000 0 1 cp -0.1000 arE2 1
HE2 CE2 1.0 1.080 120.000 180.000 0 0 h 0.1000 arE2 0
CD2 CE2 1.5 CG 1.5 1.376 118.000 0.000 0 1 cp -0.1000 arD2 1
HD2 CD2 1.0 1.080 120.000 180.000 0 0 h 0.1000 arD2 0
PHEn 21 Phenylalanine, neutral N-terminus
N N 1.0 0.000 0.000 0.000 0 0 n2 -0.5000 pepN 1
CA N 1.0 1.436 0.000 0.000 0 0 ca 0.1200 pepN 0
HN1 N 1.0 1.080 123.000 0.000 0 0 hn 0.1400 pepN 0
HN2 N 1.0 1.080 123.000 180.000 0 0 hn 0.1400 pepN 0
HA CA 1.0 1.080 110.000 120.000 0 0 h 0.1000 pepN 0
C CA 1.0 1.509 109.600 0.000 0 1 c' 0.3800 pepC 1
123456789_123456789_123456789_123456789_123456789_123456789_123456789_123456789_Starting with Version 2.2 of the Discover program, the first line of the residue library must specify the version number. If a version record is missing, an old format style is assumed. For residue libraries following the format described here, the correct version number is 2.2. The version number must be a floating-point number in columns 10-15.
2.2
123456789_123456789_123456789_123456789_123456789_123456789_123456789_123456789_
The 1- to 4- letter abbreviation for a residue (the residue name) is always found at the very beginning of the list of atoms for that residue. The next residue name follows the list of atoms for the previous residue. For example, in PHE begins the list of atoms pertaining to phenylalanine, and PHEn follows the list of atoms pertaining to phenylalanine, PHE (PHEn begins the list of atoms pertaining to the neutral N-terminus version of phenylalanine). Remember, the naming conventions are completely optional.
Columns 26-30, Number of Atoms
PHE 20 Phenylalanine, polypeptide residue
Columns 36-45, pKa values
PHE 20 Phenylalanine, polypeptide residue
Columns 47-127, Residue comments
PHE 20 Phenylalanine, polypeptide residue
Atom Cards/Second and Following Lines of a Residue
Columns 1-4, Residue Atoms
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
Columns 16-19, Ring Closure Atoms (not required by Insight II)
CD2 CE2 1.5 CG 1.5 1.376 118.000 0.000 0 1 CP -0.1000 arD2 1
Columns 21-23, Ring Closure Bond Order (may be set to 0.0 for Insight II)
CD2 CE2 1.5 CG 1.5 1.376 118.000 0.000 0 1 CP -0.1000 arD2 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
Columns 33-39, Valence Angle Parameters (may be set to 0.0 for Insight II)
CA N 1.0 1.436 123.000 180.000 omeg 0 0 ca 0.1200 pepN 0
Columns 42-48, Torsion Angle Parameters (may be set to 0.0 for Insight II)
C CA 1.0 1.509 109.600 180.000 phi 0 1 c' 0.3800 pepC 1
Columns 50-53, Torsion Angle Names
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
Column 57, Out-of-Plane Flag
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
Columns 71-74, Charge Group Name
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
N C 1.0 * 1.348 116.300 180.000 psi 0 1 n -0.5000 pepN 1
Terminal Card/EOF Last Line of Residue Library
The input for the entire residue library ends with the word elib in columns 1-4 of the final line of the file.
Specific Format for Insight's Residue Library
Insight's default residue library contains experimental data for the 20 standard amino acid residues and for other selected residues. The internal coordinate information that is contained in the residue library is meant to reflect, as much as possible, experimental structural information obtained from X-ray diffraction. Information on the original structural data is provided in Hagler et al. (1978, 1979a-c, 1985). (For complete references, refer to the Insight II references list.) For each residue, the information provided in the residue library consists of:
Capped residues are represented by an N in column 4 for an amino terminal NH3 (e.g., GLYN for NH3-glycyl-); an n in column 4 for an amide terminal NH2 (e.g., GLYn for NH2-glycyl-); or a C in column 4 for a carbonyl terminal COO- (e.g., GLYC for -glycine). Table 30 shows a summary of these residue naming conventions.
| Residue Name | Description |
|---|---|
| res | Internal neutral residue. |
| res+ | Positively charged residue. |
| res- | Negatively charged residue. |
| resN | Charged amino terminal NH3+. |
| resn | Neutral amino terminal NH2. |
| resC | Charged carboxyl terminal COO-. |
Residue Atom Names
Atom names are based on the Brookhaven naming convention, using the Greek alphabet. The backbone atoms are represented by N, CA, C, and O; then all the remaining atoms (the side chain atoms) are named in order from the alpha carbon CA. Table 31 includes examples of residue atom names and their corresponding Greek letters.
| Greek Letter |
Greek Name |
Residue Library Letter |
Examples of Atom Names |
|---|---|---|---|
| a | alpha | A | CA, HA |
| b | beta | B | CB, HB |
| g | gamma | G | CG, HG, OG, SG |
| d | delta | D | CD, HD, OD, ND, SD |
| e | epsilon | E | CE, HE, OE, NE, SE |
| z | zeta | Z | CZ, HZ, OZ, NZ |
| h | eta | H | CH, HH, OH, NH |
If there are two or more atoms in the same position relative to CA, the atoms are given number representations. For example, in PHE (), two carbons are in the delta position; they are thus designated CD1 and CD2.
Torsion Angle Names
Torsion angle names are provided for selected torsion angles. Table 32 includes the torsion angle names and their corresponding torsion angles found in Insight's residue library. Note that a torsion name is associated with the residue in which the grandparent atom is found.
| Torsional Angle Name | Atoms Included in Angle1 |
|---|---|
phi ( i) i)
| Ci-CAi-Ni-Ci-1 |
psi ( i) i)
| Ni+1-Ci-CAi-Ni |
omeg ( i) i)
| CAi-Ci-Ni+1-CAi+1 |
chi1 (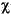 i1) i1)
| Ni-CAi-CBi-CGi |
|
chi2 (i2)
| CAi-CBi-CGi-CDi |
|
chi3 (i3)
| CBi-CGi-CDi-CEi |
| 1
Ci-1 means this atom exists in the previous residue.
|
Definitions
Parents
Grandparents
Greatgrandparents
The .rstrnt file contains descriptions of the restraints to be applied during a minimization or dynamics calculation. The .rstrnt file replaces the .noe file that was used in earlier versions of Discover. The .rstrnt file has three sections. The distance and dihedral sections specify the upper and lower bounds for applying the restraint, as well as the force constants for the biharmonic restraining force outside this range. The chiral section specifies the chirality to be achieved at asymmetric centers.
Distance, Torsion, Chiral, and NOE Volume Restraints (.rstrnt)
!BIOSYM restraint nwhere n is an integer (usually 1). Discover then interprets the file as being an ASCII file containing restraint records as outlined here.
The identifier lines introducing the sections are:
#remote_prochiral_centers #chiral #distance #NOE_distance #NOE_distance_overlapped #mixing_times #NOE_volume #NOE_volume_overlapped #NMR_dihedral #3J_dihedral
molecule#:residuename_residue#:atomnamewhere the molecule number, residue name, residue number, and atom name are as defined in the .mdf file. Colons (:) and underscores (_) are used to delimit these numbers and names as shown.
The atom name can be that of an actual atom, a pseudoatom defined in the atom set section of the .mdf file or a pseudoatom defined using the define average command in Discover.
A previously undefined pseudoatom can be referenced with wildcards or a list. Wildcards can be used for pseudoatoms consisting of atoms in the same residue if all these atoms have names beginning with some common characters. For example, if atoms 1:ASN_2:HB1 and 1:ASN_2:HB2 are present, then 1:ASN_2:HB* defines a pseudoatom consisting of these two atoms. The asterisk wildcard can match strings of any length. These two atoms can also be referred as a list, that is,
The pseudoatom is defined when the wildcard or list appears for the first time in the .rstrnt file. Thereafter, this pseudoatom is used whenever the same pattern appears.
One of a pair of prochiral hydrogens can be selected by using its prochiral specification. For example, on encountering the atom name HBS, Discover looks in the specified residue to find two atoms with names HB1 and HB2, determines their prochirality, and selects the pro-S atom to be used in the restraint. Similarly, on encountering HGR*, Discover looks for two pseudoatoms with names HG1* and HG2*, creates the pseudoatoms if necessary from (HG11,HG12,HG13) and from (HG21,HG22,HG23), and then selects the pro-R pseudoatom to use in the restraint. In each case, the character R (or S) is replaced with 1 or 2 and pro-R (or pro-S) is selected. Wildcards are allowed in this context.
Prochirality can be determined only if the molecular data file contains the priority sequence of the substituents at each prochiral center. If the prochiral atoms are not directly bonded to the prochiral center, the remote_prochiral_centers section of the restraints file should contain an entry indicating how these atoms are connected.
The contents of each record are described in the following sections and tables.
#remote_prochiral_centersThe identifier is followed by records as shown in Table 33. The atom specifications in this section should not use any prochiral specification, since that would lead to a cyclic definition.
#remote_prochiral_centers
1:VAL_8:HG1* 1:VAL_8:HG2* 1:VAL_8:CG1 1:VAL_8:CG2 1:VAL_8:CB
#chiralThe records in the chirality restraints section specify chirality around asymmetric centers, as shown in Table 34.
| field# | contents | comments |
|---|---|---|
| 1 | atom specification | the asymmetric center |
| 2 | S or R | one character representing the desired chirality at the center |
#chiral
1:THRN_1:CA S
1:ILE_35:CB S
#distanceThe distance restraints section specifies upper and lower bounds for distances between pairs of atoms, force constants, and a limit for the force, using the format shown in Table 35.
#distance
1:AR+N_1:CA 1:ASP-_3:CA 4.700 7.200 1.00 1.00 1000.000
1:PRO_2:CA 1:PHE_4:CA 4.700 7.200 1.00 1.00 1000.000
#NOE_distanceThis section contains distance restraints derived from NOE data. These restraints are the same as the restraints in the distance restraints section. However, the records in this section have an expanded format (Table 36), to contain additional data relevant to NOE analysis
#NOE_distance
!ATOM #1 ATOM #2 Distance Force Constant Max
! Lower Upper Upper Lower Upper Force
! + correction
1:CYS_3:HA 1:CYS_4:HN 2.00 3.00 3.00 1.000 1.000 1000.0
1:GLY_31:HA* 1:CYS_32:HN 3.00 5.00 4.00 1.000 1.000 1000.0
1:SER_6:HBR 1:ILE_7:HN 2.00 3.00 3.00 1.000 1.000 1000.0
1:VAL_8:HGS* 1:VAL_8:HA 3.00 5.00 4.00 1.000 1.000 1000.0
#NOE_distance_overlappedThis section contains overlapped distance restraints derived from NOE data. The first line of each of these restraints shares almost the same format as the NOE distance restraints. The only difference is that the column corresponding to the pseudo atom correction is absent in the overlapped restraint category. To assign multiple pairs of protons to the same restraint, one can put one additional pair per line with the continuation symbol "+" in the first column of succeeding lines.
Sample:
#NOE_distance_overlapped
!ATOM #1 ATOM #2 Effective Distance Force Constant Max
! Lower Upper Lower Upper Force
1:CYS_3:HA 1:CYS_4:HN 2.00 5.00 1.000 1.000 1000.0
+ 1:GLY_31:HA* 1:CYS_32:HN
+ 1:SER_6:HBR 1:ILE_7:HN
#mixing_timesEach field contains the value of the mixing times (in seconds) at which the subsequent NOE_Volume restraints were determined.
The format of each entry is as shown in Table 39.
| field# | contents | comments |
|---|---|---|
| 1 | tmix1 | mixing time 1 |
| 2 | tmix2 | mixing time 2 |
| m | tmixm | mixing time m |
#mixing_timesThe sample specifies that the subsequent volume entries are associated with mixing times of 50, 100, 150, and 200 ms.
0.05 0.1 0.15 0.2
#NOE_volumeThis section contains NOE peaks volume restraints derived from experimentally measured NOE peak volumes or integrals. In the direct NOE refinement scheme, the volume restraints are compared to theoretical NOE volumes calculated for the current model structure. The number of fields will be 2m + 4, where m is the number of mixing times.
The format of each entry is as shown in Table 36.
#NOE_volume 1:GLY_2:HAR 1:PHE_HD* 0.075 0.125 0.175 0.225 0.275 0.325 0.375 0.425 40 80
1:ALA_4:HB* 1:CYS_10:HAR -999.0 -999.0 0.175 0.225 0.275 0.325 0.375 0.425 40 80..
#NOE_volume_overlappedThis section contains NOE peaks volume restraints derived from experimentally measured overlapped NOE peak volumes or integrals. In the direct NOE refinement scheme, the volume restraints are compared to theoretical NOE volumes calculated for the current model structure. The first line of each overlapped restraint shares the same format as that of the non overlapped case. The number of fields will be 2m + 4, where m is the number of mixing times. Each succeeding line then adds a spin pair to the definition of the overlapped peaks.
The format of each entry is as shown in Table 36.
#NOE_volume 1:GLY_2:HAR 1:PHE_HD* 0.075 0.125 0.175 0.225 0.275 0.325 0.375 0.425 40 80
+ 1:ALA_4:HB* 1:CYS_10:HAR
#NMR_dihedralEach record specifies a range for a dihedral angle and the force constants for the biharmonic restraint force (Table 43).
#NMR_dihedral
1:CYS_4:C 1:PRO_5:N 1:PRO_5:CA 1:PRO_5:C -120.0 -60 50.0 50.0 500.0
#3J_dihedralEach record (see Table 44) specifies up to four ranges of dihedral angles and two force constants for the multiple-interval biharmonic restraining force. Lower and upper bounds of angles are specified for each interval. However, the same force constant is applied for all deviations from any lower bound and another force constant for all deviations from any upper bound.
!BIOSYM restraint 1
!
#remote_prochiral_centers
1:LEU_6:HD2* 1:LEU_6:HD1* 1:LEU_6:CD2 1:LEU_6:CD1 1:LEU_6:CG
!
#chiral
1:AR+N_1:CA S
1:PRO_2:CA S
!
#distance
1:AR+N_1:CA 1:ASP-_3:CA 4.700 7.200 32.00 32.00 1000.000
1:PRO_2:CA 1:PHE_4:CA 4.700 7.200 32.00 32.00 1000.000
!
#NOE_distance
!ATOM #1 ATOM #2 Distance Force Constant Max
! Lower Upper Upper Lower Upper Force
! + Correction
1:AR+N_1:HA 1:AR+N_1:HG1 -1.000 4.000 4.000 32.00 32.00 1000.000
1:MET_52:HG1 1:MET_52:HE* -1.000 5.000 4.000 32.00 32.00 1000.000
!
#NMR_dihedral
1:ASP-_3:N 1:ASP-_3:CA 1:ASP-_3:CB 1:ASP-_3:CG -120.000 0.000 30.00 30.00 1000.000
1:CYS_55:N 1:CYS_55:CA 1:CYS_55:CB 1:CYS_55:SG -120.000 0.000 30.00 30.00 1000.000
!
#3J_dihedral
1:ASP-_3:HN 1:ASP-_3:N 1:ASP-_3:CA 1:ASP-_3:HA 3.98 1.00 30.00 30.00 1000.000
1:AR+N_1:HA 1:AR+N_1:CA 1:AR+N_1:CB 1:AR+N_1:HB1 6.73 1.00 30.00 30.00 1000.000 23.5 47.9 119.4 140.3 -140.3 -119.4 -47.9 -23.5 !A=9.500, B=-1.600, C=1.800
1:PRO_2:HA 1:PRO_2:CA 1:PRO_2:CB 1:PRO_2:HB1 9.02 1.00 30.00 30.00 1000.000 131.8 153.2 -153.2 -131.8 -31.1 31.1 !A=9.500, B=-1.600, C=1.800
1:ASP-_3:HA 1:ASP-_3:CA 1:ASP-_3:CB 1:ASP-_3:HB1 12.51 1.00 30.00 30.00 1000.000 153.5 -153.5 !A=9.500, B=-1.600, C=1.800
1:PRO_8:HA 1:PRO_8:CA 1:PRO_8:CB 1:PRO_8:HB1 2.61 1.00 30.00 30.00 1000.000
1:PRO_9:HA 1:PRO_9:CA 1:PRO_9:CB 1:PRO_9:HB1 2.23 1.00 30.00 30.00 1000.000
The format of the .scs_tor files is described and illustrated with examples in the following section.
The .xdr_tor file contains the same information as the .scs_tor file, in a compact format that conserves disk space by up to 95% relative to an ASCII file. XDR files can be shared by programs running on various platforms (IRIS, IBM, ESV, etc.). A utility called scs_xdrtor is provided to inter-convert these two file types (see Appendix D, Utilities, in the Search_Compare User Guide).
The direct output of a search is an .xdr_tor file. It is not readily user-readable, but can be converted to an ASCII file by the utility scs_xdrtor. The .scs_tor file then contains the search information, including the defined rotatable bonds, the search order, implicit torsions and ring closure bonds (if rings are involved in the search), the number of atoms, the number of conformers, the anchor atom (if requested), the energy values (if generated), and the values of the torsion angles. The general format of the .scs_tor output file is similar to that of the .scs_prm input file. The .scs_tor file is described below, then illustrated by example.
!BIOSYM scs_torsion 1.2The ! must be the first character in the file. The 1.2 is the file version number. Version 1 and 1.1 files can still be read.
#rotatable_bondsAll rotatable bonds are defined by the four atoms constituting the torsion angle. This information is contained in the file in the following format:
RB# atom1 atom2 atom3 atom4RB# signifies the name of each rotatable bond, where the # indicates an integer number, and each of the four atoms defining the rotatable bonds is specified in the conventional Insight II manner:
molecule_name:residue_name:atom_name
#search_orderThis section consists of one line that lists the rotatable bonds (as RB#) in the order in which the search was performed.
#implicit_torsionsThis section exists only if the search includes ring bonds. The section consists of one line that lists the rotatable bonds (as RB#) that are implicit torsions.
#closure_bondThis section exists only if the search includes ring bonds. All closure bonds are defined by two atoms forming the ring bonds. The format of this information in the file is:
atom1 atom2The atom is specified by:
molecule_name:residue_name:atom_name
#nb_atomsThis section consists of a single number indicating the total number of atoms in the molecule.
#nb_conformersThis section consists of a single number indicating the total number of conformers found by the search. It comes before the torsion values section.
#anchor_atomThis section exists only if there is a user-specified anchor atom and gives the name of the anchor atom. It comes before the torsion values section. If an anchor atom section is not present, this means that no anchor atom was defined by you. The anchor atom name is specified in the conventional Insight manner:
molecule_name:residue_name:atom_name
#energyThis section consists only of the section keyword, which functions as a flag indicating that there is an energy value after the value of the torsion for each conformer in the torsion values section. This keyword must come before the torsion values section. The complete format is:
#energy
!
#torsion_values
angle1 angle2 ... anglen-1 number_of_values
anglen1 energyn1
anglen2 energyn2
...
#torsion_valuesThis section lists the values for the (n - 1) rotatable bonds and the number of values (number of lines that follow) for the nth rotatable bond. The format is:
angle1 angle2 ... anglen-1 number_of_values
anglen1
anglen2
...
!BIOSYM scs_torsion 1.2
!
#rotatable_bonds
RB0 CAPTO_ANALOG:1:N1 CAPTO_ANALOG:1:C8 CAPTO_ANALOG:1:C9 CAPTO_ANALOG:1:O4
RB1 CAPTO_ANALOG:1:C CAPTO_ANALOG:1:N1 CAPTO_ANALOG:1:C8 CAPTO_ANALOG:1:C9
RB2 CAPTO_ANALOG:1:C2 CAPTO_ANALOG:1:C CAPTO_ANALOG:1:N1 CAPTO_ANALOG:1:C8
RB3 CAPTO_ANALOG:1:N CAPTO_ANALOG:1:C2 CAPTO_ANALOG:1:C CAPTO_ANALOG:1:N1
!
#search_order
RB3 RB2 RB1 RB0
!
#nb_atoms
32
!
#nb_conformers
24
!
#torsion_values
-30.000000 -90.000000 -60.000000 1
60.000
150.000000 -30.000000 -60.000000 2
150.000
0.000
150.000000 -60.000000 -30.000000 1
120.000
150.000000 -60.000000 -60.000000 2
90.000
60.000
150.000000 -60.000000 90.000000 1
-150.000
150.000000 -90.000000 120.000000 1
-90.000
150.000000 -90.000000 60.000000 3
-90.000
-120.000
90.000
150.000000 -90.000000 30.000000 1
120.000
150.000000 -120.000000 90.000000 1
-60.000
150.000000 -120.000000 60.000000 1
-60.000
90.000000 -90.000000 90.000000 1
30.000
90.000000 90.000000 -60.000000 1
-90.000
90.000000 60.000000 -90.000000 2
-30.000
150.000
90.000000 60.000000 -120.000000 4
120.000
90.000
60.000
30.000
90.000000 30.000000 -90.000000 1
150.000
60.000000 30.000000 60.000000 1
-150.000
The run_name.xdr_tor or run_name.scs_tor file is used when a prior search is post-processed (as an energy or distance map). This is indicated by the presence of the keyword use_prior_search in the #scs_commands section of the .scs_prm file.
Search_Compare generates several kinds of output files:
For an explanation of the .sd format from MDL, see your MDL product documentation.
| A = ALA | G = GLY | M = MET | S = SER |
| C = CYS | H = HIS | N = ASN | T = THR |
| D = ASP | I = ILE | P = PRO | V = VAL |
| E = GLU | K = LYS | Q = GLN | W = TRP |
| F = PHE | L = LEU | R = ARG | Y = TYR |
Sample .seq File
Here is an example of an amino acid sequence in the correct format for a sequence file:
VMTQSPSSLSVSAGERVTMSCKSSQSLLNSGNQKNFLAWYQQKPGQPPKLIYGAS TRESGVPDRFTGSGSGTDFTLTISS
VQAEDLAVYYC
The format of the file consists of the "atomset" section of the molecular data file (.mdf). Refer to that section of this book for a detailed description of the file format, beginning on page 58.
!BIOSYM subset_data 4
#atomset
@degree 6 subset CRN_RINGS
CRN:PHE_13:CG CD1 CD2 CE1 CE2 CZ
CRN:TYR_29:CG CD1 CD2 CE1 CE2 CZ
CRN:TYR_44:CG CD1 CD2 CE1 CE2 CZ
#end
1.100000 2.200000 3.400000 3.500000 4.400000
1.100000 2.200000 3.400000 3.500000 4.400000
1.100000 2.200000 3.400000 3.500000 4.400000
1.100000 2.200000 3.400000 3.500000 4.400000
The top of the file is where informational text and comments may appear. There is no limit to the number of comment lines; however, when a title delimiter (#) appears as the first character in the line, function specifications must follow on the next line.
A function specification record follows each delimiter. Each field of information is preceded by its identifier in capital letters. The order of the fields must not vary from the order specified here.
#Each column of values in the file must be identified by a function specification record. The first function specification record corresponds to the first column of values, the second specification corresponds to the second column, and so on.
TITLE: Time in ps
MEASUREMENT TYPE: Time
UNITS OF MEASUREMENT: ps
FUNCTION: Time
There must be two delimiters following the last function specification record, each on a separate line. These two delimiters signify that the axis function values follow. There is no limit to the number of axis functions that may be written to a file.
The number of observables for each axis function may vary; however, the number of rows for each column must be equal. To accomplish this, columns may be padded with asterisks following the last value. The asterisks serve as placeholders, so that when the file is read, the values are associated with the correct axis function. Numeric values may not follow an asterisk in any column (i.e., holes in the data are not allowed).
When two axis functions that do not have an equal number of observables are plotted together, only the minimum number of data points result. For example, if the x axis is time and has 30 observables, and the y axis is distance and has only 25 observables (followed by 5 asterisks), the resulting plot has only 25 points.
> Bad file format
Sample 1INSIGHT V3.0 DATE: Thu Jan 25 15:50:38 1990 Sample 1 {line 1: header info}
6.0000 179.9988 0.0004
Sample 2
INSIGHT V3.0 DATE: Mon Jan 29 11:12:46 1990 Sample 2 { line 1: header info}
9.0000 16.0268
Sample 3
INSIGHT V3.0 DATE: Thu Jan 25 15:50:38 1990 Sample 3
Sample 4
INSIGHT V3.0 DATE: Mon Jan 29 11:12:46 1990 Sample 4
Sample 5
INSIGHT V3.0 DATE: Thu Jan 25 15:50:38 1990 Sample 5
Sample 6
# {Note that a file may contain as many or as few}
Sample 7
INSIGHT V3.0 DATE: Thu Jan 25 15:50:38 1990 Sample 7
Sample 8
INSIGHT V3.0 DATE: Mon Jan 29 11:12:46 1990 Sample 8
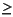 2.9.0)
2.9.0)
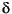 3J
3J