
| DMol |


Only one keyword and its associated options may appear on a line of the .input file. The keywords in the .input file are case-insensitive (i.e., you may use upper- and/or lowercase characters interchangeably).
Comment lines (specified by beginning the line with "#") and blank lines are ignored.
Thus, a typical .input file would look like:
# Comment
.input file keywords and options are fully described in Appendix E, Commands--Standalone Mode and summarized according to function in Chapter 7, Command Summary--Standalone Mode.
The .input file can be created with a text editor, in the dmol_master interface, or in the dialog interface. When the .input file is used, DMol automatically creates the corresponding .inmol file. The valid keywords for .input are documented in Appendix E.
Sample .input file
The following .input file shows both the file's format and the default values for all keywords.
# # These are the MSI-set defaults for the DMol UIF parameters # Product DMol Version 960 # Primary Job Keywords Calculate energy Basis dnp Geometry car angs Symmetry auto Spin restricted Charge 0.000000 Occupation default # DFT Specifications Functionals vwn Nonlocal energy Integration_Grid medium # Environment Point_Charges off Electric_Field off Solvate off # Molecular Properties Electrostatic_Moments off Mulliken_analysis 0 Hirshfeld_analysis 0 Bond_Order off ESP_Charges 0 Nuclear_EFG 0 Optical_Absorption off Plot off Grid box 3 -20 -20 -20 3.000000 # Additional Job Control Ndiff 1 Vibdif 0.010000 Project on FrqRestart 0 Lmax default Frozen inner_core Fixoc 1000 Print off Partial_DOS off # SCF Tolerances, Limits & Convergence Criteria SCF_Density_Convergence 0.000001 SCF_Energy_Convergence off SCF_Iterations 25 Mixing_Alpha 0.250000 Diis 4 Mixing_Beta 0.250000 SCF_Restart off Smear 0.000000 Number_Bad_Steps 9 Direct_SCF on # Optimizer Tolerances & Convergence Controls Locate minimum TS_Mode 0 Opt_Use_Symmetry on GDIIS off Max_Displacement 0.3000 Hessian_Update default Opt_Print 2 Opt_Coordinate_System auto Gradient_Convergence 0.001000 Displacement_Convergence 0.001000 Opt_Energy_Convergence 0.000010 Opt_Cycles 20
Example of line 6a for methylene (CH2) triplet state with one symmetry block
3 1.0 1.0 (3 doubly occupied orbitals) 2 1.0 0.0 (2 alpha-occupied orbitals) 0 (end of occupation input)See Occupation for more information on specifying occupations.
Example of lines 11 and 12 for N2, with the atoms lying on the z axis at ±1.1
3 -2. -2. -3.1 16 2. -2. -3.1 16 -2. 2. -3.1 25 -2. -2. 3.1To obtain a two-dimensional plot along the N-N bond in the yz plane, use:
2 -2. -2. -3.1 16 -2. 2. -3.1 25 -2. -2. 3.1
h2o dnp test case
3 0 2 1 1 1 1 0 0
25 0 0 5 20 0 3 0 0
0.0000000000 0.0000000000 0.0000000000 8
0.0000000000 1.2661164123 1.6440616101 1
0.0000000000 -1.2661164123 1.6440616101 1
8 10 0 0 0 0 0 0 2 2 2 2
1 5 0 0 0 2 2
0 0.00000 0 0.00000
0 0 5 5 1 0.000100 10.000000 1.000000
0.300E+00 0.300E+00 0.100E-05 0.100E-05 0.100E-05 0.100E-02
0 0 0 0 0 1 1 0 0
3 -3.7795 -3.7795 -3.7795 12 -3.7795 -3.7795 5.2912
11 -3.7795 4.5354 -3.7795 10 3.7795 -3.7795 -3.7795
The .car file is always written regardless of the input coordinate format. It is updated during geometry optimization. The starting coordinates are copied into a .car.orig file.
A utility csh script add_car_to_arc is used by the DMol executable to accomplish making .arc files. You can use this script to make .arc files for other executables as well.
| The printed energies are now binding energy instead of total energy. This change was done to make values fit in the .arc file format, so .arc files can be read into the Insight interface. |
Only standalone runs may omit an .mdf file or use another coordinate input format.
Geometry car angs
@CHARGES number of charges X Y Z CHARGE X Y Z CHARGE ...Where number of charges is an integer corresponding to the number of charges, and all fields are in floating-point format.
78.40 1082 162 1.000000 7.000000 0.200000 1.672740 0.020520
1 1.08
6 1.53
7 1.48
8 1.36
15 1.75
16 1.70
17 1.65
where 78.4 is the dielectric constant of water, 1082 is the number of the basic grid points per atom, 162 is the number of segments on the atomic surface, 1.0 is the solvent radius, 7.0 is the A-matrix cutoff, 0.2 is the radius increment, 1.672740 is the A-constant for the nonelectrostatic contributions, and 0.020520 is the B-constant. Below this line are the atomic numbers and corresponding atomic radii for several of the most commonly used elements. All the distances are in angstroms. The meaning of this information is explained under COSMO--solvation effects.
You should use 1082 basic grid points. When you do, the program tries to access an improved basic grid from the file $BIOSYM/data/dmol/cosmo.grid. If this file does not exist, the default grid is used; however, it is less homogeneous and therefore not recommended.
espfit.inp file
The espfit.inp file contains three sections:
CHARGE GRID VDWThe first section, CHARGE, contains information about the initial value of the point charge and the request to optimize or not optimize that charge. The general, free format is:
Iatom, Charge, Optimizewhere Iatom is the atom number as it appears in the run_name.car file, Charge is the value of the charge in atomic units to be optimized if the Optimize parameter is 1. If Optimize is 0 the point charge for atom Iatom is fixed during the optimization. The default is to optimize point charges for all atoms.
The CHARGE section is followed by the GRID section, which provides information as to the grid spacing and layer border diffusion (see Fitting atomic point charges to the electrostatic potential (ESP)). The default values are grid spacing: 0.3 Å; layer border diffusion: 0.1 Å.
The VDW section contains the internal and the external values of the atom shells, as follows:
Iatnum, Rint, Rextwhere Iatnum is the atomic number, and Rint and Rext are the internal and external radii, respectively, of the atom in angstroms.
The values of atomic radii as listed in this section are those commonly used in calculations of ESP-fitted charges. For atoms that are not listed in this section, the default atomic radii are reasonable; however, they have not been extensively tested.
Default espfit.inp file contents
The contents of the default $BIOSYM/data/dmol/espfit.inp file, as mentioned under ESP-fitted charges, are:
CHARGE GRID 0.3d+00 0.1d+00 VDW 1 1.45 2.80 3 2.02 2.82 6 1.50 2.80 7 1.70 2.80 8 1.70 2.80 9 1.70 2.80 11 2.31 3.11 12 2.00 2.80 14 1.65 2.80 15 2.00 2.80 16 2.00 2.80 17 1.95 2.80 19 2.67 3.47 20 2.33 3.13 30 2.08 2.80 35 2.05 2.90 37 2.81 3.61 38 2.46 3.26 48 2.31 3.11 53 2.20 3.30 55 3.01 3.81 56 2.68 3.48
The first few lines of this file are as follows:
# --------------------------------------------------------------------- # Standard Atomic Weights, Isotope Abundancies and Masses # # Version 1.0 4/18/96 # --------------------------------------------------------------------- 1 H 1.007940 ISOTOPE 1 99.9850 1.007825 ISOTOPE 2 0.0150 2.014000 ISOTOPE 3 0.0000 3.016050 ...For each atom, there is one header line containing the element number followed by the element symbol followed by the standard atomic weight.
This line is then followed by lines for every isotope of this element. The isotope lines start with the keyword ISOTOPE, followed by the number of nucleons, followed by the abundancy (as a percentage) followed by the isotope mass.
The keywords for the thermodynamic quantities enthalpy, entropy, Gibbs energy/free energy, and heat capacity (at constant pressure) are @H, @S, @G and @Cp, respectively. These keywords are followed by any of the four keywords: tra, rot, vib, and tot, which stand for translational, rotational, vibrational, and total contributions, respectively.
########################################################################
Stat_Mech Sample Input File
stat_mech.input
########################################################################
# STAT_MECH Input File generated by InsightII V4.0.0
# for Molecule <QUICK0>
@Atoms
3
8 3.99130702 0.85986608 -9.08387661 16.00000000
1 4.94494820 0.82509947 -8.83887482 1.00800002
1 3.76692677 -0.07698990 -9.29032421 1.00800002
@Frequencies
3
1626.71996157
3609.74629830
3749.73739121
@Temperature_Range
273.1500 373.1500 1.0000
@H tra rot vib tot
@S tra rot vib tot
@G tra rot vib tot
@Cp tra rot vib tot
@ZPVE
@Rotor_Info
@Symmetry
IZ X Y Zwhere IZ is the atomic number as an integer; and X, Y, and Z are the Cartesian coordinates. Terminate the input with a blank line.
To read the geometry from the .coord file while running the dmol_master interface, you can enter the character string coord when prompted for the geometry. The geometry is read from the run_name.coord file and is written in Cartesian coordinates to the .input file.
Alternatively in the .input file, the Geometry keyword can be set to a value of coord and the units to bohr or angs:
Geometry coord bohrIn this case, the geometry is extracted from the .coord file when the job runs.
This procedure is most useful when mapping a potential energy surface by repeated energy calculations. The .input file can remain the same for all geometries, and only the .coord file needs to be updated. Due to the simplicity of this file format, this is generally the most convenient way to input molecular geometries that have already been formatted in other programs.
i iz bond theta tors ib ithet itor where:Input is terminated with a blank line or with i = 0.
i The ith atom in the input.
iz Atomic number of the atom (iz = 0 indicates a
dummy atom, to be dropped from the final list).
bond Bond distance to atom ib.
theta Angle in degrees formed by atoms i, ib, and ithet
(with atom ib at the vertex).
tors Torsion angle in degrees formed by atoms i, ib,
ithet, and itor.
ib Atom number to which atom i forms a bond of
bond angstroms or Bohrs.
ithet Atom number with which atoms i and ib form an
angle of theta degrees (with atom ib at the vertex).
itor Atom number with which atoms i, ib, and ithet
form a torsion angle of tors degrees.
For i = 1, only iz is specified.
The first atom is always at the origin. The second atom always lies on the x axis, and the third atom always lies in the xy plane
To read the geometry from the .zmat file while using dmol_master, you can enter the character string zmat when prompted for the geometry. The geometry is read from the run_name.zmat file and is written in Cartesian coordinates to the .input file.
Alternatively in the .input file, the Geometry keyword can be set to the value zmat and the units to angs or bohr:
Geometry zmat angsIn this case, the geometry is extracted from the .zmat file when the job runs.
This procedure is most useful when mapping a potential energy surface by repeated energy calculations. The .input file can remain the same for all geometries, and only the .zmat file needs to be updated. The following example is a .zmat file for ferrocene:
1 26 2 0 1.695 1 3 6 1.207 90.0 2 1 4 6 1.207 72.0 90.0 2 3 1 5 6 1.207 72.0 -90.0 2 3 1 6 6 1.207 72.0 90.0 2 4 1 7 6 1.207 72.0 -90.0 2 5 1 8 0 1.000 90.0 0.0 1 2 3 9 0 1.695 90.0 180.0 1 8 2 10 6 1.207 90.0 0.0 9 1 8 11 6 1.207 72.0 90.0 9 10 1 12 6 1.207 72.0 -90.0 9 10 1 13 6 1.207 72.0 90.0 9 11 1 14 6 1.207 72.0 -90.0 9 12 1 15 1 1.076 177.2 180.0 3 2 1 16 1 1.076 177.2 180.0 4 2 1 17 1 1.076 177.2 180.0 5 2 1 18 1 1.076 177.2 180.0 6 2 1 19 1 1.076 177.2 180.0 7 2 1 20 1 1.076 177.2 180.0 10 9 1 21 1 1.076 177.2 180.0 11 9 1 22 1 1.076 177.2 180.0 12 9 1 23 1 1.076 177.2 180.0 13 9 1 24 1 1.076 177.2 180.0 14 9 1
iz ib bond ithet theta itor tors where:Input is terminated with a blank line.
iz Elemental symbol of atom (iz = X indicates a
dummy atom, to be dropped from the final list).
ib Atom number to which atom i forms a bond of
bond angstroms or Bohrs.
bond Bond distance to atom ib.
ithet Atom number with which atoms i and ib form an
angle of theta degrees (with atom ib at the vertex).
theta Angle in degrees formed by atoms i, ib, and ithet
(with atom ib at the vertex).
itor Atom number with which atoms i, ib, and ithet
form a torsion angle of tors degrees.
tors Torsion angle in degrees formed by atoms i, ib,
ithet, and itor.
For i = 1, only iz is specified.
The first atom is always at the origin, the second atom always lies on the x axis, and the third atom always lies in the xy plane.
To read in the geometry from the .gzmat file when using dmol_master, you can enter the character string gzmat when prompted for the geometry. The geometry is read from the run_name.gzmat file and is written in Cartesian coordinates to the .input file.
Alternatively in the .input file, the Geometry keyword can be set to a value of gzmat and the units to angs or bohr:
Geometry zmat angsIn this case, the geometry is extracted from the .gzmat file when the job runs.
This procedure is most useful when mapping a potential energy surface by repeated energy calculations. The .input file can remain the same for all geometries, and only the .gzmat file needs to be updated. The following example is a .gzmat file for ferrocene:
FE X 1 1.695 C 2 1.207 1 90.0 C 2 1.207 3 72.0 1 90.0 C 2 1.207 3 72.0 1 -90.0 C 2 1.207 4 72.0 1 90.0 C 2 1.207 5 72.0 1 -90.0 X 1 1.000 2 90.0 3 0.0 X 1 1.695 8 90.0 2 180.0 C 9 1.207 1 90.0 8 0.0 C 9 1.207 10 72.0 1 90.0 C 9 1.207 10 72.0 1 -90.0 C 9 1.207 11 72.0 1 90.0 C 9 1.207 12 72.0 1 -90.0 H 3 1.076 2 177.2 1 180.0 H 4 1.076 2 177.2 1 180.0 H 5 1.076 2 177.2 1 180.0 H 6 1.076 2 177.2 1 180.0 H 7 1.076 2 177.2 1 180.0 H 10 1.076 9 177.2 1 180.0 H 11 1.076 9 177.2 1 180.0 H 12 1.076 9 177.2 1 180.0 H 13 1.076 9 177.2 1 180.0 H 14 1.076 9 177.2 1 180.0
1 1 1 1 2 0
> scan_outmol run_name.outmol -lyou can obtain a summary of the final results of the DMol run. The .sum file begins with a title and a list of the most important keywords:
Product: dmol Job Name: test12 Calculation Type: Optimize Frequency Nonlocal: energy Functionals: vwn Symmetry: C1 Charge: 0.000000 Spin: restricted Basis: MIN Frozen: none Lmax: default Grid: MEDIUMNext comes a list of the optimized Cartesian coordinates and the final gradients for all atoms in the system.
============================
Optimized Molecular Geometry
============================
Cartesian Coordinates (Angstrom) Cartesian Gradients (au)
-------------------------------- -----------------------------
Atom x y z dE/dx dE/dy dE/dz
-----------
1 O 0.000000 0.000000 0.473801 0.000000 0.000000 -0.000144
2 H -0.859495 0.000000 -0.236900 0.000144 0.000000 -0.000033
3 H 0.859495 0.000000 -0.236900 -0.000144 0.000000 -0.000033
----------- -------------------------------- -----------------------------
Following this appear the total SCF energy and binding energy.
==================
Molecular Energies
==================
Electronic Energies:
--------------------
Total SCF Energy = -75.7663438 Hartree
-47544.050 kcal/mol
Final Binding Energy = -0.2815919 Hartree
-7.6625140 eV
-176.7014047 kcal/mol
-----------------------------------------------------
Next a summary of the orbital energies and occupancies is displayed.
==================
Molecular Orbitals
==================
State Eigenvalue Occ.
(au) (eV)
--------------------- ---------- ---------- -----
1 + 1 a -18.582376 -505.653 2.000
2 + 2 a -0.894501 -24.341 2.000
3 + 3 a -0.455773 -12.402 2.000
4 + 4 a -0.320333 -8.717 2.000
5 + 5 a -0.248404 -6.759 2.000
6 + 6 a -0.025379 -0.691 0.000
7 + 7 a 0.067754 1.844 0.000
--------------------- ---------- ---------- -----
In the next section the vibrational frequencies and intensities are summarized.
====================
Vibrational Spectrum
====================
mode a.u. cm-1 km/mol
---- ------ ------ ------
7 0.2854 1466.9 34.91
8 0.5454 2803.5 47.04
9 0.6013 3091.0 3.39
---- ------ ------ ------
Thermodynamic properties are listed next.
========================================================= Standard Thermodynamic Quantities at 298.15K and 1.00 Atm ========================================================= H,Trans: 0.889 kcal/mol H,Rot : 0.889 kcal/mol H,Vib : 10.527 kcal/mol S,Trans: 34.609 cal/mol.K S,Rot : 12.760 cal/mol.K S,Vib : 0.014 cal/mol.K C,Trans: 4.968 cal/mol.K C,Rot : 2.981 cal/mol.K C,Vib : 0.085 cal/mol.K H,Total: 12.305 kcal/mol S,Total: 47.382 cal/mol.K C,Total: 8.033 cal/mol.K G,Total: -1.822 kcal/mol -----------------------------------------------------If the dipole moments and Mulliken, Hirshfeld, or ESP charges are computed, they are printed as follows.
========================
Atom Centered Properties
========================
Atom Partial Charges (au)
------------ --------------------------------------
Mulliken Hirshfeld ESP
1 O -0.6824 -0.2930 -0.6112
2 H 0.3412 0.1465 0.3056
3 H 0.3412 0.1465 0.3056
------------ --------------------------------------
====================
Molecular Properties
====================
Electrostatic Moments
---------------------
Dipole Moment (Debye)
---------------------
<x>: 0.000000 <y>: 0.000000 <z>: -2.066830
2.0668 Debye
Data for the optical absorption spectra, if requested, follow:
=================================================================
Transition Dipoles <i(occ)|x,y,z|a(virt)> [au]; Alpha Orbitals
=================================================================
[ 9 Transitions with high Intensity] [Atomic Units]
--------------------------------------------------------------------------
Transition <i|x|a> <i|y|a> <i|z|a> Ea-Ei Intensity Ea-Ei
(cm-1)
1B2.1 -> 4A1.1 0.0000 0.1729 0.0000 0.2230(a) 0.03 48948.9
3A1.1 -> 4A1.1 0.0000 0.0000 0.7517 0.2950(a) 0.56 64735.8
3A1.1 -> 2B1.1 0.7773 0.0000 0.0000 0.3881(a) 0.60 85176.2
1B1.1 -> 4A1.1 1.0587 0.0000 0.0000 0.4304(a) 1.12 94461.3
1B1.1 -> 2B1.1 0.0000 0.0000 0.7594 0.5235(a) 0.58 114901.6
2A1.1 -> 4A1.1 0.0000 0.0000 0.1292 0.8691(a) 0.02 190751.3
2A1.1 -> 2B1.1 0.1509 0.0000 0.0000 0.9623(a) 0.02 211191.6
1A1.1 -> 4A1.1 0.0000 0.0000 0.0407 18.5570(a) 0.00 4072790.7
1A1.1 -> 2B1.1 0.0560 0.0000 0.0000 18.6501(a) 0.00 4093231.0
--------------------------------------------------------------------------
Finally, the job statistics are printed.
============== Job Statistics ============== Computer: iris54 Platform: irix5r4 Optimization Cycles: 5 CPU Time: 0.443
> scan_outmol run_name.outmolThis run_name.sum file contains the history of the DMol run. As before, the .sum file begins with a title and a list of the most important keywords.
Product: dmol Job Name: test12 Calculation Type: Optimize Frequency Nonlocal: energy Functionals: vwn Symmetry: C1 Charge: 0.000000 Spin: restricted Basis: MIN Frozen: none Lmax: default Grid: MEDIUMNext the history of the SCF iterations is shown. To see if a running job is converging, look at both the energies and the Convergence column. The energies should converge to a constant value, and the convergence should decrease towards zero. If the calculation is not converging, several factors could be responsible:
==================================================
Molecular Energies (for latest optimization cycle)
==================================================
E_Cycle Total Energy Binding Energy Convergence Time
------- ------------ -------------- ----------- ----
1 -75.790778 -0.306026 0.064944 0.1
2 -75.771814 -0.287062 0.018964 0.1
3 -75.766694 -0.281942 0.005120 0.1
4 -75.766424 -0.281672 0.000270 0.1
5 -75.766348 -0.281596 0.000076 0.1
6 -75.766344 -0.281592 0.000004 0.1
7 -75.766344 -0.281592 0.000000 0.1
8 -75.766344 -0.281592 0.000000 0.2
------- ------------ -------------- ----------- ----
Following this, the total SCF energy and binding energy appear.
Electronic Energies:
--------------------
Total SCF Energy = -75.7663438 Hartree
-47544.050 kcal/mol
Final Binding Energy = -0.2815919 Hartree
-7.6625140 eV
-176.7014047 kcal/mol
-----------------------------------------------------
For an optimization run, the key results from every optimization cycle are given. These are total energies, maximum gradients, displacement, and the two Hessian eigenvalues. Convergence is achieved if the energy converges to a constant and the gradient becomes smaller than a threshold, typically 0.001 au.
==================== Optimization Results ==================== Cycle Energy Gradient Displace mode1 mode2 ----- ---------- -------- -------- ----- ----- 1 -75.744379 0.147484 0.272746 0.200 0.500 2 -75.765144 0.019924 0.069463 0.203 0.469 3 -75.766273 0.007111 0.034115 0.152 0.419 4 -75.766340 0.000814 0.001903 0.159 0.394 5 -75.766344 0.000191 0.000609 0.156 0.304 ----- ---------- -------- -------- ----- -----The vibrational spectrum data are displayed next.
====================
Vibrational Spectrum
====================
mode a.u. cm-1 km/mol
---- ------ ------ ------
7 0.2854 1466.9 34.91
8 0.5454 2803.5 47.04
9 0.6013 3091.0 3.39
---- ------ ------ ------
Finally, the thermodynamic data are presented.
========================================================= Standard Thermodynamic Quantities at 298.15K and 1.00 Atm ========================================================= H,Trans: 0.889 kcal/mol H,Rot : 0.889 kcal/mol H,Vib : 10.527 kcal/mol S,Trans: 34.609 cal/mol.K S,Rot : 12.760 cal/mol.K S,Vib : 0.014 cal/mol.K C,Trans: 4.968 cal/mol.K C,Rot : 2.981 cal/mol.K C,Vib : 0.085 cal/mol.K H,Total: 12.305 kcal/mol S,Total: 47.382 cal/mol.K C,Total: 8.033 cal/mol.K G,Total: -1.822 kcal/molThis summary file is useful for monitoring convergence of both SCF and geometry calculations in DMol runs.
# geometry optimization and thermochemistry for water # Product DMol Version 960 Calculate OPTIMIZE_FREQUENCY Basis DNP Symmetry C2V Spin RESTRICTED Charge 0.000000 Functionals VWN Integration_Grid MEDIUM Frozen INNER_CORE Lmax DEFAULT Electrostatic_Moments ON Mulliken_Analysis 3The .outmol file starts with a header that gives the version number of the program and copyright information.
DMol Version 960 Density Functional Theory electronic structure program. Copyright (c) 1996 by Molecular Simulations, Inc.Following this comes a summary of all the input flags in the input file. This is the same summary that is printed out during a dmol_master or dialog session.
INPUT FLAGS:
__________________________________________________________________________
Calculate optimize_frequency Basis DNP
Geometry car angs Symmetry C2V
Spin restricted Charge 0.000000
Occupation DEFAULT
Integration_Grid MEDIUM Nonlocal energy
Functionals VWN
Electric_Field off
Point_Charges off
Solvate off
Electrostatic_Moments on Mulliken_analysis 3
Bond_Order off
Hirshfeld_analysis 0
ESP_Charges off
Nuclear_EFG off
Optical_Absorption off
Plot off
Ndiff 1 Vibdif 0.010000
Project on FrqRestart 1
Frozen inner_core Lmax default
Fixoc 1000 Print off
Smear 0.000000 SCF_Density_Convergence 1e-06
Number_Bad_Steps 9 SCF_Energy_Convergence Off
SCF_Iterations 25 Direct_SCF on
Mixing_Alpha 0.250000 Partial_DOS off
DIIS 4
Mixing_Beta 0.250000 SCF_Restart off
Locate minimum
Opt_Coordinate_System auto Opt_Print 2
Opt_Use_Symmetry on Gradient_Convergence 0.001000
Hessian_Update default Displacement_Convergence 0.001000
GDIIS off Opt_Energy_Convergence 0.000010
Opt_Cycles 20 Max_Displacement 0.300000
__________________________________________________________________________
Following this come several lines that echo these flags in .inmol-type format:
nat= 3 nspin=0 nfroz= 2 ihirsh=0 imull=3 iplot=0 idip=1 nitpri= 0 iscf= 25 iuwav=0 iupot=0 itask=4 idrct=1 nbstep= 9 idofld=0 idos=0 Molecule Rotation Matrix: 1.00000 0.00000 0.00000 0.00000 1.00000 0.00000 0.00000 0.00000 1.00000 Molecule Center Of Mass Offset: 0.00000 0.00000 0.00000These variables are described in under .inmol file; they reflect the input options that are indicated above.
specifications for basis sets: nfile= 8 nbas= 2 nfroz=10 1 0 0 0 0 0 2 2 2 2 nfile= 1 nbas= 1 nfroz= 5 0 0 0 2 2Here nfile refers to the basis set number in the .basis file (which equals the atomic number in the .basis file supplied by MSI), nbas counts the total number of basis sets that have been read in. These indicate whether an atomic basis function is ignored completely (=2), included as a valence orbital (=0), or included as a frozen core (=1). Following these lines appears a summary of the basis set information extracted from the .basis file.
Hydrogen nbas= 1 z= 1. 5 radial functions, spin energy= -0.033 n=1 L=0 occ= 1.00 e= -0.233471 -6.3531 n=1 L=0 occ= 0.00 e= -0.845000 -22.9936 n=2 L=1 occ= 0.00 e= -0.211250 -5.7484 n=2 L=1 occ= 0.00 e= -2.000000 -54.4228 eliminated n=1 L=0 occ= 0.00 e= -8.000000 -217.6912 eliminated Oxygen nbas= 2 z= 8. 10 radial functions, spin energy= -0.054 n=1 L=0 occ= 2.00 e= -18.758245 -510.4380 frozen n=2 L=0 occ= 2.00 e= -0.871362 -23.7110 n=2 L=1 occ= 4.00 e= -0.338381 -9.2078 n=2 L=0 occ= 0.00 e= -2.130332 -57.9693 n=2 L=1 occ= 0.00 e= -1.593734 -43.3677 n=3 L=2 occ= 0.00 e= -1.388889 -37.7936 n=2 L=1 occ= 0.00 e= -3.125000 -85.0356 eliminated n=1 L=0 occ= 0.00 e= -12.500000 -340.1425 eliminated n=3 L=2 occ= 0.00 e= -2.722222 -74.0755 eliminated n=2 L=1 occ= 0.00 e= -6.124999 -166.6698 eliminatedThis includes the atom name, nuclear charge, total number of basis functions (referred to as radial functions), atomic spin energy, and total atomic energy (spin-restricted). Spin energy is the difference in the atomic energies between spin-restricted and spin-unrestricted calculations. Next comes a list of each atomic orbital, or radial function, specifying the principal quantum number, angular momentum, occupation in the atomic calculation, and orbital eigenvalue in Hartrees and in eV. Last comes a flag telling how the basis function is used in the calculation. This can be frozen, meaning frozen core; blank, meaning active; or eliminated, meaning dropped from the calculation completely.
The next two lines show the density functional methods used:
vwn none none Vosko Wilk Nusair local correlationThe next section summarizes the symmetry information:
nsym= 4 ihmi= 0 icax= 4 nprs= 0 group=C2Vnsym is the number of symmetry elements in the group, ihmi = 2 if
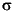 h is present in the group and 0 otherwise, icax is the size of the smallest fraction of a circle that can be used to generate all integration points, and nprs is a print option. In general, icax may be higher than the order of the principal axis.
h is present in the group and 0 otherwise, icax is the size of the smallest fraction of a circle that can be used to generate all integration points, and nprs is a print option. In general, icax may be higher than the order of the principal axis.The next section summarizes the total number of orbitals:
n norb jdegn representation
1 10 1 A1.1
2 2 1 A2.1
3 7 1 B1.1
4 4 1 B2.1
total number of valence orbitals: 23
n norc jdegn representation
1 1 1 A1.1
2 0 1 A2.1
3 0 1 B1.1
4 0 1 B2.1
total number of core orbitals: 1
need: mws, mwfm, mwvc, mwn, mwv, mwc, mwm, coef
96 169 10 25 23 1 10 4
current dimensions:
96 169 10 25 23 1 10 4
norb is the number of active orbitals and norc is the number of frozen cores. This is printed for each irreducible representation. The column jdegn lists the dimension of the representation (always 1 for Abelian groups), and the column labelled representation shows the Schönflies notation for the representation.Next comes a summary of atomic coordinates.
coordinates for atoms atom type mceq bas file element
0.000000 0.000000 0.760643 1 1 1 2 8 Oxygen
-1.466430 0.000000 -0.380322 2 2 2 1 1 Hydrogen
1.466430 0.000000 -0.380322 3 2 2 1 1 Hydrogen
The coordinates are the Cartesian coordinates in Bohrs. atom is simply a sequential list of the atoms in the order in which they appeared in the input file. type reflects the different atomic numbers of atoms in the input--each different atomic number is assigned a new type. mceq indicates which atoms are symmetrically equivalent. The column bas indicates the order in which basis sets are found in the .basis file. In this example, the hydrogen basis set is located first (bas = 1) and oxygen second (bas = 2). file corresponds to the position of the basis set in the .basis file. When you use the .basis file created with the program, this corresponds to the atomic number.Several sections appear that show input data, including the orbital occupations, parameters for the integration procedure, and the maximum angular momentum of the fitting functions. The occupation information looks like:
occup input as read:
0 0.00000 1000 0.00000
as interpreted:
iopt= 0 0.00 icfr= 1000 delte= 4.97E-09
molecule charge= 0.0 active electron number= 8.0
including core= 10.0 (without charge= 10.0)
Setting iopt = 0 tells the program to attempt to determine the optimal orbital occupation. Setting icfr = 1000 instructs the program to attempt this for the first 1000 iterations, which should be all iterations. iopt can be controlled by changing the .inmol input or in .input with the Occupation keyword.Next, information for the fitting basis is read. This indicates the number of spherical harmonic functions that are used in the analytic representation of the model density and electrostatic potential:
prolo input as read from SYMDEC:
16 0 0 1 0 ! nrf npr mlod ipart iref
as interpreted:
nrf,npr,mlod,ipart,iref 16 0 0 1 0
nrf,mwf 12 18
modef 62427 500000 563 9
This output indicates that 16 multipolar functions are used and that partition function type 1 is used to partition the model density. Following this comes information about the number of gradient directions to be evaluated:
prede1 input as read from SYMDEC:
3 0
as interpreted:
modes,nprder 3 0
The number of gradients is 3, corresponding to oxygen z and symmetric hydrogen z and y displacements. nprder is a print option, set to zero in this case. modef is currently disabled.The section controlling the numerical integration parameters appears as follows:
parti3 input as read:
0 0 5 5 1 0.000100 10.000000 1.000000
as interpreted:
inputs,npri,ipa,iomax,iomin,thres,rmaxp,sp
0 0 5 5 1 0.00010 10.00000 1.00000
wta dimension 4282
file type nrtb zn rmaxp thres thresh iomax iomin lmaxv lmaxz
8 1 30 8. 10.000 0.00010 0.00000333 5 2 3 2
1 2 20 1. 10.000 0.00010 0.00000500 5 1 2 1
Integration points and checksum: 1276 9.999975
Integration points and checksum: 1276 9.999975
mwp 4276
Memory use data:
nloop= 3509 3510 312 328 98581
nloopd= 3005 3006 192 192 9 10
int array elements available (maxi): 500000 ( 1.9 Mb)
real array elements available (maxr): 1250000 ( 9.5 Mb)
minimum real array elements needed: 107630 ( 0.8 Mb)
real array elements used: 672302 ( 5.1 Mb)
The value of 0 for inputs indicates that no data are input for special atoms--the same defaults are used in all cases. (The print option npri can currently be set only in an .inmol input file.) These control the quality of the integration mesh. On the next line, the total number of mesh points is printed, along with the sum of the atomic densities computed on this mesh. The value shown here is in error by only 6E-5, which is quite good.Next follows a number of parameters that control the SCF calculation:
SCF parameter input as read: 2.500E-01 2.500E-01 1.000E-06 0.000E+00 as interpreted: mixing parameters 0.25000 0.25000 Density tolerance for converging SCF: 1.000E-06 smallest core norm at, symtry, function 1.000074 1 1Next begin the actual self-consistent iterations for solution of the LDF equations.
A summary of the results from each iteration of the self-consistent procedure appears after each iteration. This information includes:
Total Energy Binding E Cnvgnce_Dens Cnvgnce_E Time
ef -0.7597604855E+02 -0.4912966 0.0466342 - 0.0
ef -0.7590937881E+02 -0.4246269 0.0138991 0.06666974 0.0
ef -0.7590105792E+02 -0.4163060 0.0100172 0.00832089 0.0
ef -0.7589813699E+02 -0.4133851 0.0020630 0.00292093 0.0
ef -0.7589799202E+02 -0.4132401 0.0003130 0.00014497 0.1
ef -0.7589796290E+02 -0.4132110 0.0001410 0.00002912 0.1
ef -0.7589796185E+02 -0.4132099 0.0000070 0.00000104 0.1
ef -0.7589796175E+02 -0.4132098 0.0000049 0.00000010 0.1
ef -0.7589796175E+02 -0.4132098 0.0000004 0.00000000 0.1
This output is discussed in detail above, for the .sum file. The string ef is a convenience for performing a search on the output file. If too many print options have been enabled, these data may be difficult to see, since the eigenvectors and eigenvalues may also be printed out each iteration (these are omitted from this example).If requested, a derivative is performed next. The data are summarized in atomic units:
df ATOMIC COORDINATES DERIVATIVES df x y z x y z df O 0.000000 0.000000 0.760643 0.000000 0.000000 0.001356 df H -1.466430 0.000000 -0.380322 -0.000067 0.000000 0.000710 df H 1.466430 0.000000 -0.380322 0.000067 0.000000 0.000710 en total energy: -75.8979617 au -2065.28950 eV -47626.609 Kcal/mol en binding energy: -0.4132098 au -11.24402 eV -259.293 Kcal/mol en nuclear repulsion energy: 8.9523072 auFollowing the final iteration appear the MO eigenvalues in Hartrees and the orbital occupations; these appear in columns, one for each molecular orbital. Expansion coefficients, if requested, appear only for active (not frozen) orbitals. MOs are grouped first by atom, and then by angular momentum, as illustrated in the following example:
Eigenvalues and occupations:
Alpha orbitals, symmetry block 1 A1.1
Degeneracy: 1 Size: 10
-0.92050E+00 -0.34677E+00 -0.18087E-01 0.86639E-01 0.17483E+00 0.53301E+00 0.69993E+00 0.83403E+00 0.10619E+01 0.16849E+01
2.00 2.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
Alpha orbitals, symmetry block 2 A2.1
Degeneracy: 1 Size: 2
0.17973E+00 0.93957E+00
0.00 0.00
Alpha orbitals, symmetry block 3 B1.1
Degeneracy: 1 Size: 7
-0.48549E+00 0.35010E-01 0.15682E+00 0.22545E+00 0.70146E+00 0.10736E+01 0.21747E+01
2.00 0.00 0.00 0.00 0.00 0.00 0.00
Alpha orbitals, symmetry block 4 B2.1
Degeneracy: 1 Size: 4
-0.26857E+00 0.79979E-01 0.71363E+00 0.82303E+00
2.00 0.00 0.00 0.00
A record of the geometry optimization follows:
** GEOMETRY OPTIMIZATION IN INTERNAL COORDINATES **
Searching for a Minimum
Optimization Cycle: 1
Coordinates (Angstroms)
ATOM X Y Z
1 o 0.000000 0.000000 0.402515
2 h -0.776001 0.000000 -0.201258
3 h 0.776001 0.000000 -0.201258
Point Group: C2V Number of degrees of freedom: 2
Energy is -75.897961749
2 Hessian modes will be used to form the next step
Hessian Eigenvalues:
0.200000 0.500000
Minimum Search - Taking Simple RFO Step
Searching for Lamda that Minimizes Along All modes
Value Taken Lamda = -0.00000042
Step Taken. Stepsize is 0.001310
Maximum Tolerance Cnvgd?
Gradient 0.000262 0.001000 YES
Displacement 0.001201 0.001000 NO
Energy change --- 0.000010 NO
opt (E=) -75.8979617 (G=) 0.000262 (D=) 0.001201
New Cartesian Coordinates Obtained by Inverse Iteration
Cycle: 1 Maximum deviation: 0.00120075
Cycle: 2 Maximum deviation: 0.00000047
Cycle: 3 Maximum deviation: 0.00000000
Displacement from previous Coordinates is: 0.001061
Total Energy Binding E Cnvgnce_Dens Cnvgnce_E Time
Integration points and checksum: 1276 9.999975
Integration points and checksum: 1276 9.999975
smallest core norm at, symtry, function 1.000074 1 1
ef -0.7589795988E+02 -0.4132080 0.0000272 - 0.1
ef -0.7589796019E+02 -0.4132083 0.0000100 0.00000031 0.1
ef -0.7589796026E+02 -0.4132083 0.0000053 0.00000007 0.1
ef -0.7589796019E+02 -0.4132083 0.0000004 0.00000007 0.1
df ATOMIC COORDINATES DERIVATIVES
df x y z x y z
df O 0.000000 0.000000 0.759905 0.000000 0.000000 0.001037
df H -1.466823 0.000000 -0.379952 -0.000013 0.000000 0.000872
df H 1.466823 0.000000 -0.379952 0.000013 0.000000 0.000872
en total energy: -75.8979602 au -2065.28946 eV -47626.608 Kcal/mol
en binding energy: -0.4132083 au -11.24397 eV -259.292 Kcal/mol
en nuclear repulsion energy: 8.9539305 au
Cartesian Hessian Update
Hessian Updated using BFGS Update
** GEOMETRY OPTIMIZATION IN INTERNAL COORDINATES **
Searching for a Minimum
Optimization Cycle: 2 ...
The molecular orbital spectrum appears next:
Molecular orbital spectrum:
energy of Highest Occupied Molecular Orbital -0.268577 -7.308
number of eigenvalues listed: 23
state eigenvalue occupation
(au) (ev)
1 + 1 A1.1 -0.920530 -25.049 2.000
2 + 1 B1.1 -0.485682 -13.216 2.000
3 + 2 A1.1 -0.346655 -9.433 2.000
4 + 1 B2.1 -0.268577 -7.308 2.000 ...
20 + 9 A1.1 1.061992 28.898 0.000
21 + 6 B1.1 1.073702 29.217 0.000
22 + 10 A1.1 1.684908 45.849 0.000
23 + 7 B1.1 2.175225 59.191 0.000
The dipole moment appears next, if its evaluation is requested:
Dipole moment vectors (au):
x y z
electronic: 0.000000000 0.000000000 -4.530077247
nuclear: 0.000000000 0.000000000 3.799524158
net: 0.000000000 0.000000000 -0.730553089
dipole magnitude: 0.73055308873 au
1.85689792435 debye
If requested, the population analysis begins in the next section. If the population type requested is 2 or 3, the first result displayed is the population matrix (=3) or the reduced population matrix (=2). This gives the effective number of electrons in terms of overlapping pairs of atomic orbitals. For the population matrix, this sum extends over all active atomic orbitals in the basis set. For a reduced population matrix, the sum extends over the types of orbitals in the basis (s, p, d, etc.). The population matrices are followed by an analysis that essentially sums the columns of the population matrices. These give the effective occupations for atomic orbitals, followed by a reduced population analysis for orbital types. Each row contains the atom name and its number as input in the geometry. Following this is an index that indicates the atomic orbital quantum numbers n, l, and ml. Finally come the orbital occupation numbers for 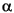 - and
- and 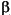 -spin orbitals; their sum appears in the column labeled charge and their difference in the spin column.
-spin orbitals; their sum appears in the column labeled charge and their difference in the spin column.
Mulliken Population analysis
Population matrices are scaled by 1000. for display
Population analysis for representation 1 a
1 20 0 1.812 1740.
1 20 0 -0.010 0. 4.
1 21-1 1.933 0. 0. 1890.
1 21-1 0.007 0. 0. 0. 4.
1 21 0 1.617 0. 0. 0. 0. 1398.
1 21 0 0.001 0. 0. 0. 0. 0. 2.
1 21 1 1.472 0. 0. 0. 0. 0. 0. 1216.
1 21 1 0.005 0. 0. 0. 0. 0. 0. 0. 1.
1 32-2 0.000 0. 0. 0. 0. 0. 0. 0. 0. 0.
1 32-1 0.006 0. 0. 0. 0. 0. 0. 0. 0. 0. 6.
1 32 0 0.008 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 6.
1 32 1 0.038 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 11.
1 32 2 0.002 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1.
2 10 0 0.348 30. -9. 0. 0. 83. -1. 65. 2. 0. 0. 0. 5. 1. 137.
2 10 0 0.130 -8. 2. 0. 0. 20. 0. 50. 0. 0. 0. 0. 8. 0. 0. 69.
2 21-1 0.026 0. 0. 22. 2. 0. 0. 0. 0. 0. 1. 0. 0. 0. 0. 0. 2.
2 21 0 0.012 5. 0. 0. 0. 7. 0. -1. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1.
2 21 1 0.034 9. 0. 0. 0. 1. 0. 15. 1. 0. 0. 0. 1. 0. 0. 0. 0. 0. 3.
3 10 0 0.348 30. -9. 0. 0. 83. -1. 65. 2. 0. 0. 0. 5. 1. 28. 0. 0. 0. 8. 137.
3 10 0 0.130 -8. 2. 0. 0. 20. 0. 50. 0. 0. 0. 0. 8. 0. 0. -8. 0. 0. -3. 0.
69.
3 21-1 0.026 0. 0. 22. 2. 0. 0. 0. 0. 0. 1. 0. 0. 0. 0. 0. 1. 0. 0. 0.
0. 2.
3 21 0 0.012 5. 0. 0. 0. 7. 0. -1. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0.
0. 0. 1.
3 21 1 0.034 9. 0. 0. 0. 1. 0. 15. 1. 0. 0. 0. 1. 0. 8. -3. 0. 0. 1. 0.
0. 0. 0. 3.
spin up down charge spin
O( 1) 20 0 0.9060 0.9060 1.8121 0.0000
O( 1) 20 0 -0.0051 -0.0051 -0.0101 0.0000
O( 1) 21-1 0.9665 0.9665 1.9330 0.0000 ...
H( 3) 10 0 0.0653 0.0653 0.1306 0.0000
H( 3) 21-1 0.0132 0.0132 0.0264 0.0000
H( 3) 21 0 0.0064 0.0064 0.0128 0.0000
H( 3) 21 1 0.0174 0.0174 0.0348 0.0000
condensed population analysis
spin up down charge spin
O( 1) 20 0.9010 0.9010 1.8020 0.0000
O( 1) 21 2.5185 2.5185 5.0369 0.0000
O( 1) 32 0.0278 0.0278 0.0556 0.0000
H( 2) 10 0.2394 0.2394 0.4787 0.0000
H( 2) 21 0.0370 0.0370 0.0740 0.0000
H( 3) 10 0.2394 0.2394 0.4787 0.0000
H( 3) 21 0.0370 0.0370 0.0740 0.0000
Mulliken atomic charges:
O 1 -0.8945
H 2 0.4473
H 3 0.4473
Next come optional sections such as vibrational data, thermodynamic results, and optical absorption spectra. These should be self explanatory. For example:
Harmonic frequencies will be computed by finite differences.
Number of displacements per atom is: 1
Step size for finite differences is: 0.010000 Bohrs
finite difference step for:
atom number 1
coordinate 1
step number 1
Integration points and checksum: 4115 9.999978
Integration points and checksum: 4115 9.999978
smallest core norm at, symtry, function 1.000076 1 1
Total Energy Binding E Cnvgnce_Dens Cnvgnce_E Time
ef -0.7589794672E+02 -0.4131948 0.0002132 0.00000000 0.5
ef -0.7589794605E+02 -0.4131941 0.0001482 0.00000067 0.5
ef -0.7589794601E+02 -0.4131941 0.0000246 0.00000004 0.5
ef -0.7589794593E+02 -0.4131940 0.0000020 0.00000008 0.6
ef -0.7589794596E+02 -0.4131940 0.0000006 0.00000003 0.6
Dipole moment vectors (au):
x y z
electronic: -0.065338854 0.000000000 -4.530158647
nuclear: 0.060000000 0.000000000 3.799524158
net: -0.005338854 0.000000000 -0.730634489
dipole magnitude: 0.73065399469 au
1.85715440409 debye
df ATOMIC COORDINATES DERIVATIVES
df x y z x y z
df O 0.010000 0.000000 0.759905 0.005685 0.000000 0.001181
df H -1.466823 0.000000 -0.379952 -0.003131 0.000000 -0.001532
df H 1.466823 0.000000 -0.379952 -0.003139 0.000000 0.003198
finite difference step for:
atom number 1
coordinate 2
step number 1 ...
...
hessian:
0.5685307E+00
-.9200000E-05 -.2984700E-02
0.7188850E-02 0.5229500E-03 0.4148981E+00
-.2905413E+00 -.3994000E-03 -.1701898E+00 0.3429663E+00
0.6409500E-03 0.9383000E-03 0.1804500E-03 -.5365000E-03 -.2210000E-04
-.2288350E+00 -.4491000E-03 -.2004691E+00 0.2070463E+00 -.4003000E-03
0.1869308E+00
-.3071367E+00 0.4093000E-03 0.1778562E+00 -.2642910E-01 -.1160000E-04
0.3295555E-01 0.3368018E+00
-.6318000E-03 0.9396000E-03 0.1836000E-03 0.7800000E-05 -.3626000E-03
0.1690000E-04 0.5302500E-03 -.2450000E-04
0.2249323E+00 -.4552500E-03 -.2004691E+00 -.3390555E-01 0.2110000E-04
0.8774250E-02 -.2102494E+00 -.3985000E-03 0.1869308E+00
dipole moment derivatives:
0.5338854E+00 0.0000000E+00 0.8140000E-02
-.3140000E-03 0.6394306E+00 -.1524600E-02
0.2100000E-05 -.3030000E-03 0.3693550E+00
-.2675467E+00 0.0000000E+00 0.8087570E-01
0.4166000E-03 -.3197269E+00 -.1336100E-02
0.4074980E-01 0.1808000E-03 -.1780179E+00
-.2685752E+00 -.6000000E-06 -.7356890E-01
0.7132000E-03 -.3197247E+00 -.2673000E-03
-.4075170E-01 0.1819000E-03 -.1780198E+00
****************************************
vibrational frequencies, intensities
mode a.u. cm-1 km/mol
7 0.3063 1574.6 77.88
8 0.6996 3596.4 3.64
9 0.7270 3737.2 81.95
*****************************************
Frequencies (cm-1) and normal modes (ang)
7: 1574.6 8: 3596.4 9: 3737.2
O x -0.0006 -0.0007 -0.1428
y 0.0000 0.0000 0.0000
z -0.1461 -0.0999 0.0011
H x -0.2100 0.3102 0.2839
y 0.0000 0.0000 0.0000
z 0.2920 0.2000 0.2189
H x 0.2124 -0.3076 0.2850
y 0.0000 0.0000 0.0000
z 0.2902 0.1980 -0.2232
*****************************************
Dipole derivatives wrt normal modes
mode dMu/dmode [a.u.]
7: -0.0016 0.0002 -0.2820
8: -0.0008 0.0002 -0.0610
9: -0.2892 0.0000 0.0048
STANDARD THERMODYNAMIC QUANTITIES AT 298.15 K AND 1.00 ATM
Zero point vibrational energy: 12.735 kcal/mol
Atom 1 Element O Has Mass 15.99940
Atom 2 Element H Has Mass 1.00790
Atom 3 Element H Has Mass 1.00790
Molecular Mass: 18.015200 amu
Principal axes and moments of inertia in atomic units:
1 2 3
Eigenvalues -- 2.32602 4.33713 6.66315
X 1.00000 0.00000 0.00000
Y 0.00000 0.00000 1.00000
Z 0.00000 1.00000 0.00000
Rotational Symmetry Number is 1
The Molecule is an Asymetric Top
Symmetry Point Group C2V
H,Trans: 0.889 kcal/mol
H,Rot : 0.889 kcal/mol
H,Vib : 12.737 kcal/mol
S,Trans: 34.609 cal/mol.K
S,Rot : 11.984 cal/mol.K
S,Vib : 0.009 cal/mol.K
C,Trans: 4.968 cal/mol.K
C,Rot : 2.981 cal/mol.K
C,Vib : 0.058 cal/mol.K
===================================
H,Total: 14.515 kcal/mol
S,Total: 46.601 cal/mol.K
C,Total: 8.006 cal/mol.K
G,Total: 0.620 kcal/mol
_______________________________________________________________
DMol calculates the thermodynamic properties in the temperature range 198.15-298.15 K, every 10 K. The valuds of S, Cp, H, and G are listed as a function of temperature.
STANDARD THERMODYNAMIC QUANTITIES AT TEMPERATURE, T (K)
T Entropy Heat_Capacity Enthalphy Free_Energy
(K) S (cal/mol.K) C H (kcal/mol) G
________________________________________________________________
1 198.15 43.345 7.952 13.916 5.327
2 208.15 43.737 7.953 13.976 4.872
3 218.15 44.110 7.955 14.035 4.413
4 228.15 44.467 7.958 14.095 3.950 ...
15 338.15 47.612 8.059 14.756 -1.344
16 348.15 47.847 8.075 14.817 -1.841
17 358.15 48.076 8.092 14.878 -2.340
18 368.15 48.299 8.110 14.939 -2.842
19 378.15 48.517 8.129 15.001 -3.346
20 388.15 48.729 8.148 15.062 -3.852
21 398.15 48.937 8.169 15.124 -4.361
_______________________________________________________________
Finally, the .outmol file concludes with the run time:
all done time 2.514
A run using an HSOH molecule is used to illustrate the file. DMol was run using the VWN potential with the minimal (MIN) basis set. The relevant sections of the run_name.outmol file are as follows:
------------------------------------------------------------------------
Section for fitting the atomic charges to reproduce the
electrostatic potential (ESP)
------------------------------------------------------------------------
File espfit.inp does not exist and therefore the program will use
default values to build the ESP layer.
The ESP layer will be built using the following data:
The distance between points in cubic grid (A): 0.30
The width of ESP layer diffusion border (A): 0.10
atomic number and symbol internal and external radius (A)
8 O 1.70 2.80
1 H 1.45 2.80
16 S 2.00 2.80
1 H 1.45 2.80
Parameters of box for ESP calculations [A]:
Min: Max: Spaces:
1) x direction: 0.6000 9.0000 28
2) y direction: -3.0000 5.1000 27
3) z direction: -12.6000 -5.1000 25
Plotting input:
0 0 0 0 1 0 0 0 0
0: representation ordered specification of wavefcns
output is:
1 static_potential
grid specifications 3 1.134 -5.669 -23.811 25 1.134 -5.669 -9.638
grid specifications 27 1.134 9.638 -23.811 28 17.008 -5.669 -23.811
21112 grid points were specified
DMol ESP potential from the file:
run_name_potenesp.grd
will be used to calculate ESP-fitted charges.
Total number of grid points in box: 21112
Approx. weighted no. of points used: 4954
Atomic charges fit to the electrostatic potential:
(fix = charge frozen, opt = charge optimized)
Atom ESP-fitted charges:
O 1 opt -0.3747
H 2 opt 0.3192
S 3 opt -0.0431
H 4 opt 0.0987
Analysis of the ESP charge fitting results
Weighted number of used grid points: 4954.9
Range of used potential: -27.41 to 38.53 kcal/mol
RMS of used potential : 10.04 kcal/mol
RMS of fit deviation : 2.71 kcal/mol
RRMS of fit deviation : 27.0 %
ESP_fit time 2.014
----------
Explanation
First, you are informed that an espfit.inp file does not exist and therefore DMol will use default values to build the ESP layer. The input data used to build the ESP layer is listed next, followed by the parameters of the box.
A special ESP potential file is created in your current directory with the name run_name_potenesp.grd.
.cosmo file
Similar to ordinary DMol runs, several files are written during DMol/COSMO runs. Only the run_name.outmol file differs from the standard (gas phase) .outmol file. Also, an additional file that contains the COSMO results (run_name.cosmo) is created.
DMol/COSMO Results
COSMO input
Dielectric Constant = 35.90
Basic Grid Size = 1082
Number of Segments = 162
Solvent Radius = 0.30
A - Matrix Cutoff = 10.00
Radius Increment = 0.10
Non-Electrostatic Energy = A+B*area
A = 1.94242 B = 0.01515
Total energy (au) [TE]
{including COSMO solvation energy, Eq.2} = -113.6425037629
Dielectric energy (au) {1/2<q|U>} [DE] = -0.0114457888
Total energy corrected (au) [TE(corr)]
{TE(corr) = TE + DE(corr) - DE} = -113.6427402387
Dielectric energy corrected (au) [DE(corr)]
{DE(corr) = (q+q")A(q+q")} = -0.0116822646
{U is the potential on the inner cavity
it depends on the accurate density}
Sum of polarization charges {q} = -0.03270
Sum of polarization charges(corrected)
{q+q"} = -0.01282
Total surface area (Angstrom**2) = 48.03750
Nonelectrostatic Solvation Energy (kcal/mol) = 2.6701880525
Total energy corrected (au) [TE(corr)]
+ Non-Electrostatic Energy = -113.6384863825
The meanings of these terms are explained under COSMO--solvation effects.
Segment information:
n - segment number
atom - atom associated with segment n
position - segment coordinates
charge - segment charge
area - segment area
potential - solute potential on a segment
The positions of the segment (or screening charges) coordinates are given in atomic units, and the segment area is in square angstroms. The potential is computed as BQ (see Eq. 79). A partial printout of the .cosmo file follows.
n atom position (X, Y, Z) [au] charge area charge/area potential 1 1 -1.00629 2.98137 -0.98905 -0.00018 0.17784 -0.00102 0.00804 2 1 -2.06157 -2.57472 -0.21063 -0.00045 0.10670 -0.00424 0.02457 3 1 3.01776 -0.95827 -0.92618 -0.00113 0.14227 -0.00797 0.01037 4 1 2.15013 2.50419 0.23425 -0.00118 0.14227 -0.00829 0.03352 5 1 -0.52942 2.27232 2.37204 -0.00087 0.17784 -0.00487 0.05326 ... 223 4 -3.62541 -1.01677 2.01547 -0.00444 0.41810 -0.01062 0.05367 224 4 -3.65214 -0.44226 -0.11210 -0.00420 0.35121 -0.01196 0.03459 225 4 -3.30843 1.58814 0.52771 -0.00386 0.33448 -0.01154 0.04187 226 4 -2.97984 1.32927 2.53749 -0.00310 0.36793 -0.00842 0.05558 227 4 -2.79358 -0.71663 3.03613 -0.00335 0.38466 -0.00870 0.05776 228 4 -2.97706 -1.93600 1.26244 -0.00372 0.36793 -0.01011 0.04827 229 4 -2.14487 1.93148 2.26391 -0.00338 0.31776 -0.01065 0.05537 230 4 -1.76656 1.11525 3.09975 -0.00415 0.38466 -0.01080 0.05974 231 4 -1.55853 -0.11920 3.37680 -0.00394 0.36793 -0.01070 0.06092 232 4 -1.76367 -1.25666 3.01283 -0.00403 0.36793 -0.01095 0.05941 233 4 -2.18916 -2.03683 2.04514 -0.00296 0.25086 -0.01180 0.05398
Occupation Filein the input file. The program then expects the run_name.occup file to reside in the working directory. The occupancy provided in the .occup file is used throughout the DMol run.
The format and content of the .occup file are the same as for the nonstandard input of occupancy, as explained under Occupation.
Optical absorption results
The results of the optical absorption calculation are contained in the run_name.outmol and run_name.sum files and in a separate file called run_name.optabs that is used for visualization of the optical absorption spectrum with the Spectrum_Display command of the DMol module in Insight.
Plot files
The grid data for plotting each electronic property appear in individual files, which are compatible with the Contour utility of the Insight program. The files used to store data are shown in Table 6.
Each file begins with a title, which is the same as that used in the .inmol or .input file.
, , and  . The unit cell is the size of the grid used for plotting. This is generally a cube with edges parallel to the x, y, and z axes. In this case, a, b, and c define the size of the plotting grid, and the cell angles are all 90°.
. The unit cell is the size of the grid used for plotting. This is generally a cube with edges parallel to the x, y, and z axes. In this case, a, b, and c define the size of the plotting grid, and the cell angles are all 90°.
grid specifications 3 -7.30 -1.00 -5.50 15 -7.30 -1.00 5.50 grid specifications 4 -7.30 1.00 -5.50 10 4.80 -1.00 -5.50This defines a 3D grid extending from the point (-7.3x, -1.0y, -5.5z) to (+4.8x, +1.0y, +5.5z). A total of 10 spaces are used in the x direction, 4 in y, and 15 in z. The .spindens file contains the spin density, that is, the charge density for alpha electrons minus that for beta electrons. It looks like:
Hydrazene calculation with d,d extended basis, L=3,2 potential
(1F15.10)
6.403 1.058 5.821 90.000 90.000 90.000
10 4 15
1 -6 4 -2 2 -8 7
0.0005725281
0.0006603023
0.0007598884
0.0008720257
...
0.0001487091
0.0001053702
0.0000727264
The data are the values of the spin density generated on the grid. The first line is a title. The second line is the format of the grid data. The third line is the size of the plotting grid in angstroms. For example, since x runs from -7.3 to +4.8 Bohrs (a distance of 12.1 Bohrs), the a parameter on line 3 is 6.403 Å. The fourth line reflects the grid spacings defined in the grid specifications: 10, 4, and 15 spaces in the x, y, and z directions, respectively.The fifth line indicates to the Contour utility which index moves fastest and where the origin is relative to the grid. The 1 indicates that x varies the most rapidly. Note that x is also the last direction specified in the grid specifications. The data -6 4 -2 2 -8 7 tell the program that of the 10 spaces in x, 6 lie to the left and 4 to the right of the origin. This is not quite correct, since the distance is not exactly divisible by 10. DMol writes out a coordinate file with appropriately shifted values, to be compatible with the plotting files. (See the documentation for the Insight program for more information.)
The next lines contain the actual values of the spin densities. In each direction, there is one more grid POINT than there are grid SPACES. The total number of grid points in this case is (10 + 1) (4 + 1) (15 + 1) = 880.
Because the x direction is the last specified in the grid specifications, the x coordinate varies the most rapidly. Similarly, z varies the slowest, since it is specified first. The first of the 880 data points corresponds to the value of the spin density at the origin, (-7.3x, -1.0y, -5.5z). The second data point corresponds to a coordinate displaced in the x direction (because x varies the fastest). The point is 1/10th of the way to +4.8x (because 10 spaces were specified in the input). The eleventh data point corresponds to the coordinates (+4.8x, -1.0y, -5.5z). The twelfth point restarts at -7.3x but is displaced in the y direction. Following this pattern, the distribution of the first 55 points is shown in Figure 4.
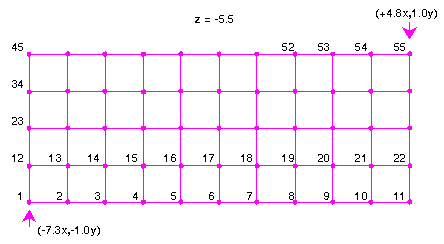
|
The 56th point begins a new z plane, displaced by 1/15th of the distance from -5.5z to +5.5z, which is shown in Figure 5.

|
This pattern continues until the final plane, z = +5.5 is reached. The last 55 points (826 through 880) correspond to this plane and are shown in Figure 6.
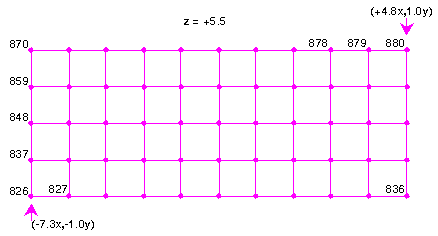
|
.hess File
The .hess file stores the gradient data generated by finite-difference calculations. With this file, it is possible to restart an interrupted frequency calculation with the keyword FrqRestart. The .hess file starts with the gradients and dipole moment for the unperturbed geometry, that is, the input geometry if a frequency calculation is requested or the optimized geometry if Optimize_Frequency is requested. Following this are the values for the gradients and dipole moments at the displaced geometries:
where:Following this come one line of input for each atom, with the gradients in Hartrees Bohr-1 (3f20.9), then the x, y and z dipole moments (in atomic units).
iat Number of atom being displaced (0 for initial
geometry).
ix Cartesian coordinate being displaced:
0 for initial geometry
1 = x
2 = y
3 = z
istep 1 for + step. The first step in 2-point difference,
or the only step of 1-point.
2 for (-) step. Second step of 2-point difference.
vibdif Size of step in finite difference.
########################################################################
Stat_Mech Sample Output File
stat_mech.output
########################################################################
# ------------------------------------------------------------------------
# STAT_MECH OUTPUT
# 273.150 K <= T <= 373.150 K. Stepsize = 1.000 K
# ------------------------------------------------------------------------
Moments of Inertia + Rotational Constants
0.66126877 1.21227439 1.87354316 [a.m.u.*A^2]
1.09808004 2.01306088 3.11114092 [10^(-40)g*cm^2]
25.49284936 13.90578359 8.99772453 [cm^(-1)]
Rotor_Info : The Molecule is an Asymetric Top
===> Symmetry Number = 2
===> Zero Point Vibration Energy [kcal/mol] = 12.846
@Temperature [K] 273.150
@Pressure [atm] 1.000
trans rot vib total
@Enthalpy [kcal/mol] 0.814 0.814 12.847 14.476
@Entropy [cal/mol.K] 34.174 10.363 0.004 44.540
@Gibbs Energy [kcal/mol] -8.520 -2.016 12.846 2.310
@Heat Capacity [cal/mol.K] 4.968 2.981 0.028 7.977
@Temperature [K] 274.150
@Pressure [atm] 1.000
trans rot vib total
@Enthalpy [kcal/mol] 0.817 0.817 12.847 14.482
@Entropy [cal/mol.K] 34.192 10.373 0.004 44.569
@Gibbs Energy [kcal/mol] -8.557 -2.027 12.846 2.263
@Heat Capacity [cal/mol.K] 4.968 2.981 0.028 7.977
@Temperature [K] 275.150
@Pressure [atm] 1.000
trans rot vib total
@Enthalpy [kcal/mol] 0.820 0.820 12.847 14.488
...
Sample stat_mech.tbl file
######################################################################## Stat_Mech Sample Table File stat_mech.tbl ######################################################################## Statistical Thermodynamics (TBL) # TITLE: Temperature MEASUREMENT TYPE: UNITS OF MEASUREMENT: K FUNCTION: Temperature # TITLE: Pressure MEASUREMENT TYPE: UNITS OF MEASUREMENT: atm FUNCTION: Pressure # TITLE: Translational Enthalpy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Translational Enthalpy # TITLE: Rotational Enthalpy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Rotational Enthalpy # TITLE: Vibrational Enthalpy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Vibrational Enthalpy # TITLE: Enthalpy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Enthalpy # TITLE: Translational Entropy MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Translational Entropy # TITLE: Rotational Entropy MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Rotational Entropy # TITLE: Vibrational Entropy MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Vibrational Entropy # TITLE: Entropy MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Entropy # TITLE: Translational Gibbs Energy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Translational Gibbs Energy # TITLE: Rotational Gibbs Energy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Rotational Gibbs Energy # TITLE: Vibrational Gibbs Energy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Vibrational Gibbs Energy # TITLE: Gibbs Energy MEASUREMENT TYPE: UNITS OF MEASUREMENT: kcal/mol FUNCTION: Gibbs Energy # TITLE: Translational Heat Capacity MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Translational Heat Capacity # TITLE: Rotational Heat Capacity MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Rotational Heat Capacity # TITLE: Vibrational Heat Capacity MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Vibrational Heat Capacity # TITLE: Heat Capacity MEASUREMENT TYPE: UNITS OF MEASUREMENT: cal/mol.K FUNCTION: Heat Capacity # # 273.150000 1.000000 0.814202 0.814202 12.847304 14.475709 34.174005 10.362593 0.003613 44.540211 -8.520427 -2.016340 12.846317 2.309550 4.967981 2.980788 0.027736 7.976505 ...
| The data for each temperature are written on one line for each temperature point. |
Scratch files
DMol creates several scratch files: