
5
Methodology--Insight

This section is a general description of how to use the DMol module of the Insight program. The on-line help utility contains further details about what individual commands and parameters do, and Tutorial--The Insight Environment describes examples of their use. On-line help (accessed by clicking the help icon) also presents information about parameters and commands.
Commands provided in the DMol module are discussed in general terms in the order in which they are used in a typical calculation. Also included are hints about when certain parameter values should or should not be used.
Much of this information is background material for the DMol tutorial lessons described under Tutorial--The Insight Environment, so you should read this section before starting them.

Using DMol in the Insight environment
The DMol module is used to set up and then start a DMol calculation. Unlike many commands in the Insight package, nothing actually happens until the DMol job is started, making it possible to set up complex calculations and even (by temporarily leaving the Insight environment to use a text editor) edit or change parts of the calculation before the job is started.
The commands in the DMol module can be grouped in four classes, roughly corresponding to the four basic steps of a DMol calculation:
The relation of these pulldowns to the basic steps of a DMol calculation is outlined in the following section, and a detailed description of how the commands and parameters accessed via these pulldowns are used is presented under Setting up calculations with the commands in the DMol module.
Outline of basic steps of a DMol calculation
Step 1: Defining the model
A DMol calculation can be performed on any molecule or assembly of molecules that exists in the Insight space (provided it is not too large). Molecules can be read from files with the File/Import or Molecule/Get command, be created with the Builder module, or result from work performed with another product such as the Discover program. Note that DMol does not modify the molecule in any way at this stage. Therefore, any desired modifications such as adding hydrogens or partial charges to a molecule or adjusting its configuration must be done prior to starting the DMol calculation. After you start DMol by selecting DMol from the Module pulldown (by clicking the MSI logo), you then tell it what system you want to work on and what basic type of calculation you want to do with the Setup/System command.
Next, the Symmetry/Find_Pt_Group command may be executed to determine the point-group symmetry of the system. Roughly symmetrical molecules can be geometrically adjusted to the exact symmetry required for DMol calculations by increasing the value of the Symmetry_Threshold parameter.
Step 2: Setting up the calculation parameters
The next step is to set the desired input parameters. The Setup/Parameters command and the Optimize pulldown contain commands that specify these parameters as well as the input and output files. Since all these parameters have default values, you need only set them if you want values other than the defaults. The internal default values for the parameters can be customized by changing the file dmol_defaults0.input. Each time you execute one of these commands, the parameter values that you enter become the new defaults and are used in all subsequent calculations that are set up in the current Insight session. You need to specify what output you want before you start a run.
You can read in parameters that were used in a previous calculation by toggling the Load_Input parameter in the Setup/System command to on. You should also set the Calculation_Type to the same type of calculation specified in the input file you load, since it will otherwise be overwritten by your new choice.
Step 3: Performing the DMol calculation
After all the input parameters are specified, you are ready to run the DMol calculation using the commands in the Background_Job and Run pulldowns.
The Background_Job pulldown is used to set nondefault run conditions.
The Run/Run_DMol command writes out the necessary input files and specifies a name that is used as the root name for all output files.
The Run_DMol command also creates a shell script file (run_name.csh) that actually runs the DMol calculation. You can have the Insight program automatically submit this job to the background or just write it to disk, so you can submit it manually later.
Step 4: Analyzing the results
The run_name.outmol file contains the results of your DMol calculation. You can view its contents with the Insight file/text viewer or any standard UNIX text reader or editor. You can also use commands in the Analyze pulldown and in other modules of the Insight interface to construct grids and graph various properties.
Setting up calculations with the commands in the DMol module
Beginning a DMol session
To access the DMol commands, select DMol from the Module pulldown (that is, click the MSI logo, then choose DMol from the list that appears). The set of DMol pulldowns then appears on the lower menu bar.
DMol uses a defaults file for setting up default values for the DMol interface in Insight. The Insight program looks in a cascading series of directories for a defaults file: first in current-working-directory, then in $HOME, and, if the file has still not been found, finally in $BIOSYM/data/dmol. You can customize the defaults file to tailor the parameter values to your liking. Site administrators may choose to update the file in $BIOSYM/data/dmol, and ordinary users will probably want to copy or modify a file in their home directory.
You can reload the DMol interface with data from a previous job. This can speed revisions when only a minor change is needed for another calculation. This is accomplished by toggling the Setup/System command's Load_Input parameter to on.
Defining the molecule and its point-group symmetry
Specifying the system and type of calculation
You use the Setup/System command to tell DMol the name of the molecule or assembly on which you want to perform calculations, as well as the basic type of calculation you want to perform.
To specify the system, enter a name in the Object Name parameter box by picking a molecule or assembly in the display area of the screen, choosing its name from the Assem/Mol Names value-aid, or typing it in the Object Name parameter box.
Select Energy to compute the electronic energy of your system. To compute energy and a single gradient without optimization, select Gradient. Select the Optimize parameter to optimize the geometry of your system. To compute harmonic frequencies at the specified geometry, select the Frequency parameter. The Optimize_Frequency parameter allows you to optimize the geometry before computing the harmonic frequencies
In addition, you can toggle the Load_Input parameter on to read in previously set parameter values from a command input file, by entering its name in the File Name parameter box. You also need to set the Calculation_Type to match that in the input file.
Finding and adjusting the point-group symmetry
Important
Toggling the Snap_Symmetry parameter on means to attempt to change the relationship of the atoms to their ideal symmetry positions (the symmetry is adjusted when the command is executed). The symmetry label is displayed whether Snap_Symmetry is on or off.
Snap_Orientation can be toggled if Snap_Symmetry is on. Toggling the Snap_Orientation parameter on means to attempt to place the molecule in the standard orientation around the origin and symmetry axes. Note: this may significantly displace your molecule. If Snap_Orientation is off, the molecule orientation does not change.
If you want to execute the Symmetry/Find_Pt_Group command with Snap_Orientation toggled on, you must do so before using the Grid pulldown, to prevent a mismatch between the positions of the molecule and the grid.
If the Symmetry/Find_Pt_Group command is not executed with Snap_Symmetry and Snap_Orientation toggled on, C1 symmetry is used in an optimization calculation (even if the Use_Symmetry parameter in the Optimize/Parameters command is toggled on).
The Find_Pt_Group command can be used in two different ways. First, you may want to use it to simply determine if there is essentially exact symmetry (other than C1, which is no symmetry) at the current geometry. This is done by executing the command once with Symmetry_Threshold set to a small value (e.g., 10-6) and the Snap parameters toggled on. The molecule is translated and/or rotated so that its center of mass resides at the origin of the Cartesian coordinate system, and any non-Abelian symmetry found is used during the subsequent DMol background job.
You should avoid setting Symmetry_Threshold to values 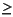 0.8-1.0 Bohr. If you find that you need to use threshold values this large, you should probably use the Builder tools to manually adjust the geometry.
0.8-1.0 Bohr. If you find that you need to use threshold values this large, you should probably use the Builder tools to manually adjust the geometry.
Specifying parameters that control the calculation
The Setup/Parameters command and the Optimize pulldown are used to define the DMol calculation parameters and the input and output files. You typically select one or more of these commands to set up the calculation conditions. The Optimize pulldown controls parameters for jobs that use the OPTIMIZE routine, and the Setup/Parameters command controls parameters that are specific to DMol.
Each of these commands contains a group of parameters that must be defined before performing a DMol calculation if you want values other than the defaults.
Using the Setup/Parameters command
The Setup/Parameters parameter block contains many parameters that allow you to control the details of the DMol calculation. (Each time you execute the Setup/Parameters command, the parameter values that you enter become the new defaults and are used in all subsequent calculations in the current Insight session.) Each of these groups of parameters is discussed separately below.
The usual default appearance of DMol's Setup/Parameters parameter block is shown below. Additional parameters may become accessible as you set various options in this parameter block.

Choosing functionals
The Functionals parameters in the Setup/Parameters command allow you to select the appropriate DFT functionals for the run.
As described under NLSD functionals in DMol, DMol supports several commonly used functionals. By default, the VWN functional is selected. This is the most popular local spin density (LSD) functional. It can be used successfully to predict molecular structures in molecules with covalent bonds. For molecules with weak (e.g., hydrogen) bonds or the obtain quantitative thermochemical results, the gradient-corrected functionals should be used. DMol supports the Becke (1988) gradient-corrected exchange functional (B_88), together with local correlation (BVWN) and the gradient-corrected correlation functionals of Perdew and Wang (1992) (BPW) and Lee, Yang, and Parr (BLYP). The gradient-corrected functionals are computationally more expensive than the local functional, but they are necessary to obtain accurate results.
If any of the gradient-corrected functionals is selected, you can set the Non_Local_Type parameter to Perturbed_Energy or SCF_Potential as the type of calculation. The Perturbed_Energy type of calculation is computationally less expensive than the SCF_Potential type. Choosing Perturbed_Energy means that the local (VWN) functional is used in SCF iterations and the final LSD density is applied for NLSD calculations of both energy and gradients. Selecting SCF_Potential means that the chosen NLSD functional is used in SCF calculations as well as in the energy and gradient calculations. This is the most accurate type of method for obtaining the new NLSD density. Experience shows, however, that the difference between the LSD and NLSD densities is rather small; therefore, the Perturbed_Energy type is recommended as a good compromise between accuracy and computational expense.
DMol allows you to select Other_Functionals, both Local and Gradient-Corrected. You can, for example, select the HL_JMW local functional instead of the VWN functional. (The JMW function was the default in previous releases of DMol.) The Other_Functionals choice is recommended for comparison or validation purposes only. In particular, caution is needed if only a correlation-gradient functional is selected, but no B_88 exchange contribution is chosen.
Parameters controlling the basis set
The Basis and Grid parameters in the Setup/Parameters command allow you to select the atomic basis functions that DMol uses in the expansion of the molecular orbitals.
To define the number and type of atomic orbitals used by DMol to expand the molecular orbitals, use the Atomic_Basis_Set parameter. Its value can be set to Minimal, Double_Numeric, DND, or DNP. The Minimal basis set uses one atomic orbital for each occupied orbital in the free atom. Minimal basis sets are generally inadequate for anything but qualitative results. The Double_Numeric basis set uses approximately two atomic orbitals for each occupied orbital in the free atom. The DNP basis set uses double-numerical basis functions together with polarization functions, i.e., functions with angular momentum one higher than that of the highest occupied orbital in the free atom. For example, the polarization function for H is 2p, for C is 3d, and for Fe is 4p. This basis set is comparable in quality to Gaussian 6-31G** sets. DNP basis functions usually yield the most reliable calculations. The DND basis set is identical to DNP except that no p functions are used for hydrogen. This basis set is comparable in size to Gaussian 6-31G* basis sets, but is of much better quality.
The input .basis file included with DMol contains at least a DNP basis set for each atom up to U. Triple-zeta basis sets and/or diffuse orbitals are also provided for some elements. Refer to the Basis set summary in Utilities for a complete list of the basis functions included for each element. Although the orbital data for all four basis sets is read from the .basis file, only the data for the basis set selected by this command are used in the DMol calculation. Atomic orbitals associated with the remaining basis sets are labeled eliminated in the output file for that run.
The Frozen_Orbitals parameter allows you to freeze specific orbitals (i.e., eliminate them from the calculation). The available parameter values are:
The Integration_Grid parameter allows you to control the selection of mesh points for the numerical integration procedure used to evaluate the matrix elements of Eqs. 17 and 18. Your choice of mesh points can significantly affect the final results of some calculations, such as electronic energy and dipole moment. You can select one of the following mesh point parameters for your calculation:
Electronic state and environment parameters
You may want to specify nondefault parameters that define the electronic state of the molecule. The Spin parameter allows you to select spin-restricted or spin-unrestricted wavefunctions. For closed-shell systems select Restricted, which forces both alpha and beta electrons into the same orbitals. Choose Unrestricted for open-shell systems to allow electrons with different spins to use different orbitals.
Although DMol automatically chooses appropriate occupations for a neutral or ionic system, you can define the total molecular charge for ionic systems using the Charge parameter. Alternatively, you can input user-specified occupations via a file called run_name.occup. These occupations remain fixed during the DMol run. Toggle User_Occupations on and input a filename for the Occup_File_Name parameter.
Toggle the Impose_Elec_Fld parameter on to impose a static electric field on the system. Setting the Impose_Elec_Fld parameter on activates the Stat_Electric_Fld parameter, which allows you to enter the X, Y, and Z coordinates, in atomic units, of the static electric field.
Set the Point_Charges parameter on to include point charges in the calculation. Setting the Point_Charges parameter on activates the Charges_File_Name parameter The Charges_File_Name parameter is filled in with the name of the point-charge file. Choose the desired file from the value-aid that appears when you click the Charges_File_Name parameter box.
To request calculations in a solvent using the COSMO model, toggle the Solvate parameter on and input the name of the cosmo_input file as the COSMO_File_Name parameter.
SCF parameters
The SCF parameters in the Setup/Parameters command allow you to define parameters for the self-consistent field (SCF) calculation.
Toggle the Restart_SCF parameter on to restart an interrupted or unconverged calculation from the .tpotl file, which contains the analytic representation of the Coulombic potential. Alternatively, using a previous .tpotl file at a different geometry can give the calculation a much better starting guess than the default, which is simply the density from the superposition of atomic densities. Setting the Restart_SCF parameter on makes the Restart_SCF_File parameter accessible, which allows you to select which .tpotl file to use.
Set the Direct_Procedure parameter on to store the SCF orbital values in memory instead of on disk. The DMol integration requires the evaluation of quantities of the form:
Eq. 130
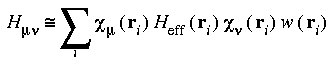 as discussed in Theory and Implementation. These calculations are usually accomplished by storing the value of each atomic orbital
as discussed in Theory and Implementation. These calculations are usually accomplished by storing the value of each atomic orbital 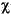 µ at each of the DMol integration grid points ri in the FWV file during each iteration of the SCF procedure. Since a medium-sized molecule may have about 300 atomic orbitals and 50,000 grid points, the FWV file can consume up to 120 megabytes of disk space.
µ at each of the DMol integration grid points ri in the FWV file during each iteration of the SCF procedure. Since a medium-sized molecule may have about 300 atomic orbitals and 50,000 grid points, the FWV file can consume up to 120 megabytes of disk space.
If disk space is a limiting factor, DMol can construct these quantities in memory as needed, rather than reading them from the disk, using what is referred to as the direct procedure. This technique has the advantage of eliminating the FWV file, thus significantly reducing disk requirements. However, there is some CPU overhead associated with this procedure. Typical calculations require 25-50% more time when you specify the direct method.
Set the Direct_Procedure parameter off to store the SCF orbital values on disk.
Toggle the SCF_Parameters parameter on to access the Iteration parameter, which allows you to specify the maximum number of iterations in the self-consistent solution for the LDF equations. Enter a larger number to achieve better convergence. However, each iteration increases the processing time. If the program exceeds this number of iterations before meeting the specified SCF convergence threshold, it automatically suspends the calculation.
Use the SCF_Dens parameter to specify the minimum degree of convergence for the SCF procedure. The degree of convergence is measured by the rms change in the density from one iteration to the next. The program automatically suspends calculation when convergence, as defined by this parameter, is reached.
The SCF procedure is terminated when the change in the total energy from one iteration to the next is less than the SCF_Ener threshold. SCF_Ener is zero (off) by default, which means that some other criterion for SCF convergence is applied. By default, the norm of the electron density matrix (SCF_Dens, see above) is used to monitor and stop the SCF process.
DIIS_Size invokes the DIIS algorithm of Pulay (Pulay, 1982). DIIS calculates the new fitted density as a linear combination of the current fitted density and densities from the previous SCF cycles. The total number of SCF cycles considered for DIIS interpolation is DIIS_Size. The default and recommended value is 4, the maximum is 10. It may be necessary to use a larger DIIS space (e.g., 8) for metallic clusters, together with small values of the Mix parameters and a non-zero value for Smearing (below).
The Mix_Alpha and Mix_Beta parameters restrict the maximum allowed change in the charge (or spin) density for each iteration of the self-consistent procedure. If 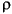 old is the charge density from the previous iteration and curr is the density from the current iteration, then the new density is calculated as:
old is the charge density from the previous iteration and curr is the density from the current iteration, then the new density is calculated as:
Eq. 131
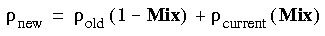 This method results in smoother convergence. During this procedure, the program actually uses several previous densities and performs an interpolation. If the interpolation appears too nonlinear, the program automatically reduces the value of the Mix parameter during the next iteration.
This method results in smoother convergence. During this procedure, the program actually uses several previous densities and performs an interpolation. If the interpolation appears too nonlinear, the program automatically reduces the value of the Mix parameter during the next iteration.
To prevent automatic reduction of the Mix parameters, specify negative values for them. (The absolute value of the parameters are actually used for the calculation.) Mix parameter values as small as 0.005 are not unusual for systems with a large number of low-lying excited states, such as metal clusters. The program also automatically reduces this parameter any time it detects an oscillatory pattern instead of convergence during the LDF self-consistent procedure.
The Bad_Steps parameter allows you to define the maximum number of consecutive bad (divergent) iterations allowed before DMol aborts an LDF calculation. Divergence may occur if the Mix parameters are too large. Since the LDF algorithm often makes a single divergent step and recovers in the next iteration, do not enter a value of 1. This option ensures that the CPU does not waste valuable time attempting to complete a divergent calculation.
The Smearing parameter allows electrons to be smeared out among all orbitals within a given 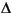 E of the Fermi level. This procedure improves convergence of the SCF procedure by allowing orbital relaxation to take place more rapidly. The character of the resulting virtual orbitals can be more effectively mixed into the occupied space, since they are now fractionally occupied. It is important to confirm that, as the density converges to its self-consistent value, the amount of charge smearing also tends to zero, to ensure that the calculation converges to the ground state.
E of the Fermi level. This procedure improves convergence of the SCF procedure by allowing orbital relaxation to take place more rapidly. The character of the resulting virtual orbitals can be more effectively mixed into the occupied space, since they are now fractionally occupied. It is important to confirm that, as the density converges to its self-consistent value, the amount of charge smearing also tends to zero, to ensure that the calculation converges to the ground state.
Large values of the Smearing parameter may prevent the calculation from converging. If this occurs, restart the calculation with the Restart_SCF option toggled on and reduce the Smearing parameter.
To smear charges within X Hartrees of the Fermi level, enter X as the Smearing parameter, where X is a floating-point number from 0.001 to 0.1. To ensure consistent results, the value of Smearing should be less than the Mix parameters by a factor of 2-10.
Properties and population analysis parameters
The Properties and Population Analysis parameters of the Setup/Parameters command allow you to specify various properties to be calculated.
Electric moments
Toggle the Electric_Moments parameter on to calculate the dipole moment.
Electric field gradients
If Nuclear_EFG is toggled on, the electric field gradients at all nuclei are calculated. For a geometry optimization, the EFG calculation is performed only for the final, optimized geometry.
Technical notes for EFG
Partial densities of states
Toggle the Partial_DOS parameter on to calculate partial density-of-states results.
Optical absorption spectra
The Optical Absorption and related parameters allow you to specify calculation of an optical absorption spectrum. Toggle the Optical Absorption option on to include optical absorption in your calculation. This also gives access to two additional parameters, which specify the energy range for which transitions are to be calculated. The energies are specified in units of wavenumbers. The default range extends between 10,000 and 250,000 cm-1.
It is possible to run optical absorption calculations with frozen-core basis sets, and for both spin restricted and spin unrestricted models.
Mulliken population analysis
You can toggle the Mulliken_Charges parameter on to enable a simple Mulliken analysis. Here, the total effective number of electrons in each atomic orbital is included in the population analysis. Toggling Mulliken_Charges to on activates the Overlap_Matrix parameter. Toggle the Overlap_Matrix parameter on to also include the population-overlap matrix in the analysis. Toggling Overlap_Matrix to on activates the Reduced_Orbital parameter. Set the Reduced_Orbital parameter on to limit the Mulliken analysis of the overlap matrix to reduced or condensed orbitals.
Bond order
Toggling the Bond_Order parameter on allows you to calculate Mulliken and Mayer bond orders and Mayer valencies as defined under Mulliken and Mayer bond orders. If the model has an odd number of electrons, then the free valence is calculated as well.
Technical note for bond
order
This option works only for models without symmetry.
Hirshfeld charges
Hirshfeld_Charges properties are determined by Atomic_Dipoles and Atomic_Quadrupoles. The Hirshfeld partitioned charges are defined relative to the deformation density. The deformation density is the difference between the molecular and the unrelaxed atomic charge densities and is given by:
Eq. 132
 where (r) is the molecular charge density and (r - R
where (r) is the molecular charge density and (r - R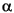 ) is the density of the free atom at coordinates R. Using the deformation density, the effective atomic charges, dipoles, and quadrupoles on atom are defined as (Delley 1986):
) is the density of the free atom at coordinates R. Using the deformation density, the effective atomic charges, dipoles, and quadrupoles on atom are defined as (Delley 1986):
Eq. 133

Eq. 134

Eq. 135
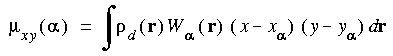 The weight W(r) is defined as the fraction of the atomic density from atom at coordinate r:
The weight W(r) is defined as the fraction of the atomic density from atom at coordinate r:
Eq. 136
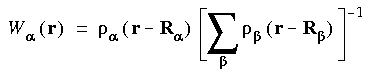 Toggle the Hirshfeld_Charges parameter on to compute effective atomic charges via the Hirshfeld partitioning procedure. Setting Hirshfeld_Charges to on also activates the Atomic_Dipoles parameter. Toggle the Atomic_Dipoles parameter on to compute the effective atomic dipoles in addition to the atomic charges. This also activates the Atomic_Quadrupoles parameter. Toggle the Atomic_Quadrupoles parameter on to also calculate the effective atomic quadrupoles.
Toggle the Hirshfeld_Charges parameter on to compute effective atomic charges via the Hirshfeld partitioning procedure. Setting Hirshfeld_Charges to on also activates the Atomic_Dipoles parameter. Toggle the Atomic_Dipoles parameter on to compute the effective atomic dipoles in addition to the atomic charges. This also activates the Atomic_Quadrupoles parameter. Toggle the Atomic_Quadrupoles parameter on to also calculate the effective atomic quadrupoles.
ESP-fitted charges
After you toggle the ESP_Charges parameter on and submit the DMol job, the run_name.input file contains the line:
ESP_Charges 1
The ESP charges are calculated at the end of the DMol run and are summarized, together with the Mulliken and Hirshfeld charges, in the summary file called run_name.sum.
The ESP charges can be displayed by using the DMol module's Analyze/Charge_Display command, as described under Displaying charges.
COSMO--solvation effects
Specifying COSMO
parameters
The Environment/COSMO command gives you access to parameters that are used to define the DMol/COSMO calculation and the input file. Each parameter in this command gives access an additional group of parameters that must be defined before performing a DMol/COSMO calculation if you want values other than the defaults.
After you select an appropriate parameter you may want to write COSMO input by toggling the Write_COSMO_Input parameter on. When this parameter is toggled on, a COSMO_Input_File parameter box appears with the default name of the input file, run_name.cosinp. You may change this name if you want. Select Execute to write the current COSMO parameter values to the COSMO_Input_File. You will then select this file in the Setup/Parameters command as described below (Specifying a COSMO calculation).
Each of the COSMO_Options is discussed separately here.
- The Dielectric_Constant parameter is used to specify the dielectric constant of the solvent. The default dielectric constant is 78.4, which corresponds to water at room temperature. You can also set COSMO_Values to Infinite for an "infinite" dielectric constant, which corresponds to a conductor. (In actuality, a value of 999.0 is used rather than infinity.) Setting COSMO_Values to By_Solvent displays a list of several common solvents. Choosing one of these solvents sets the Dielectric_Constant to the dielectric constant of that solvent. Selecting MAIN_OPTIONS returns you to the previous lists of parameters. You can enter your own value for the dielectric constant by setting COSMO_Values to User and entering a real number in the Dielectric_Constant parameter box.
- The Surface_Definition parameter allows for custom design of the cavity (i.e., the solvent-accessible) surface, which is constructed by the method of Klamt. The main parameters governing surface construction are the number of points in the basic grid, the number of segments on the surface, and the solvent probe radius.
- Typically, you do not change the values of the surface parameters, but rather leave them at their default values. The values of the default parameters are displayed in the information area near the bottom of the main Insight window. If you set COSMO_Values to User, you may change the parameters that define the surface construction. After selecting User, the allowed values of some of the parameters (as you select them from the upper list in the parameter block) are displayed under COSMO_Values. For other parameters, suggested default values (which you can change) are displayed in a Parameter_Value parameter box if you in addition set COSMO_Values to User. (This is easier to understand if you select the Environment/COSMO command in the DMol module of the Insight program and try choosing the various parameters as you read.)
- The following parameters can be specified from the Insight interface:
- We recommend that you not change the default values of surface definition parameters for typical DMol/COSMO runs.
- The Non_Electrostatic parameter controls two other parameters, A_Constant and B_Constant (accessible when you select Non_Electrostatic and then set COSMO_Values to User), which are used to approximate the nonelectrostatic contributions to the free solvation energy within the COSMO model. The two parameters define this contribution via Eq. 83, where the surface_area is the area of the cavity surface enclosing the molecule.
- The Atomic_Radii parameter is used to define the atomic radii of the atoms present in the molecule. You can modify the value of an atomic radius by setting COSMO_Values to User and then selecting the element from the COSMO_Elements list (this list is empty if you did not previously specify a molecule with the Setup/System command). The VDW_Radius parameter shows the van der Waals radius of the chosen element. You can modify the VDW_Radius by entering a different value.
Caution
|
Be cautious in changing the Atomic_Radii or Radius_Increment parameters, since COSMO results depend strongly on the choice of the size of the cavity enclosing the molecule. The A_Constant and B_Constant that define the Non_Electrostatic term were optimized for the default values of the van der Waals radii, so that changing atomic radii without reoptimization of these constants will introduce an error.
|
Specifying a COSMO calculation
Once you have written a COSMO input file by executing (either in your current Insight session or previously) the Environment/COSMO command with the Write_COSMO_Input parameter toggled on, you can specify a COSMO calculation.
Select the Setup/Parameters command and toggle the Solvate parameter on. This makes a COSMO_File_Name parameter box become accessible. You can enter the name of the COSMO input file by choosing its name from a DMol_Files value-aid. All the input COSMO files (suffix = .cosinp) that reside in the current directory appear in this list. Once you specify the COSMO input file in the COSMO_File_Name parameter box, you can set other parameters and then select Execute and proceed as usual with DMol job submission. The output of the DMol/COSMO run and the results file run_name.cosmo are described in Files.
Vibrational parameters
The Vibrational parameters in the Setup/Parameters command allow you to define vibrational parameters for a DMol calculation. These parameters are accessible if a frequency calculation has been specified with the Setup/System command.
The Finite_Differences parameter allows you to select the number of finite differences of gradients used during frequency evaluation, either Single_Point and Two_Point. Harmonic vibrational frequencies are computed by diagonalizing the mass-weighted second-derivative matrix F (Wilson et al. 1980). The elements of F are given by:
Eq. 137
 Here, qi and qj represent two Cartesian coordinates of two atoms with masses mi and mj. The square roots of the eigenvalues of F are the harmonic frequencies.
Here, qi and qj represent two Cartesian coordinates of two atoms with masses mi and mj. The square roots of the eigenvalues of F are the harmonic frequencies.
Since DMol does not calculate the second derivatives analytically, it calculates finite differences of the first derivatives instead. Gradients are computed at displaced geometries and the second derivatives are computed numerically.
Eq. 138

- The two terms represent the analytic derivatives at the equilibrium geometry and at a geometry with coordinate qj displaced by distance .
Eq. 139

- Although two-point differencing reduces numerical rounding-off, it almost doubles the computation time.
The Step_Size parameter allows you to define the step size used in the finite-difference evaluation of the Hessian. You may need to use large values for very flat potential surfaces, while small values work well for steep surfaces.
The Restart_A parameter allows you to restart a frequency evaluation starting at a specific atom. DMol calculates the gradient for each atom perturbed in the x, y, and z directions and stores it in the .hess file. If a calculation is interrupted, you can restart it beginning with displacements on atom #n, thereby saving all previous calculations. A value of 0 or 1 restarts the calculation from the first atom. A restart value greater than the number of atoms causes the program to read in all the available data in HESS and evaluate the vibrational frequencies.
The Project_Matrix parameter allows you to project the translations and rotations from the force-constant matrix any time that the force-constant (i.e., second-derivative) matrix is not evaluated at the point of the zero gradient. Instead of the 6 normal modes with value zero, corresponding to 3 translational and 3 rotational degrees of freedom, this projection forces six 0 eigenvalues. This method has a slight effect on the remaining 3N - 6 vibrational frequencies.
Using OPTIMIZE in the Insight environment
The recommended procedure for geometry optimization within the Insight program is:
- Construct the molecule to be optimized using the Builder or read in the geometry from an existing .car file or appropriate database.
- Specify any desired geometric constraints with the Optimize/Constraints command. This may include standard distance, angle, or dihedral constraints among any atoms in the system, as well as frozen Cartesian coordinates. Dummy atoms can be defined with the Pseudo_Atom pulldown (Builder module) to aid in defining constraints that are awkward or impossible to define with only real atoms.
- Minimize the geometry interactively, using one of the molecular mechanics forcefields available as part of the Discover package (the default forcefield is adequate for most routine organic molecules). It is strongly recommended that you also calculate a Hessian matrix at this time. (Starting with even a relatively simple mechanics Hessian can dramatically reduce the number of cycles required to reach convergence, especially with Cartesian coordinates.) You should also make sure that the desired constraints are met, as much as possible, before starting the quantum optimization run.
- Check the mechanics-converged geometry for symmetry using the Symmetry/Find_Pt_Group command. If no symmetry is found and you suspect that your molecule ought to have some, increase the symmetry tolerance and try again. If symmetry is still not found, try repeating Step 3 with a tighter gradient-convergence criterion.
- Steps 1-4 can be repeated to build, say, the backbone of a molecule and ensure that it has the desired symmetry and/or constraints before adding side chains and additional functional groups. (Note, that if you do modify the molecule, you will need to recalculate the Hessian, using the same mechanics forcefield as in Step 3. Do this by invoking the Discover program again.)
- When you are satisfied with your molecule's symmetry and constraints, only then should you set the remaining optimization parameters and prepare your job for submission.
The advantages of starting an optimization with a reliable Hessian are clear from Table 1, which shows the number of cycles to reach convergence with various Hessian options for a test suite of molecules covering a range of various point-group symmetries in both Cartesian and natural internal coordinates.
Using a good starting Hessian is virtually mandatory in optimizations in Cartesian space, which perform very poorly otherwise, especially for larger systems with little or no symmetry (Table 1). Optimizations in natural internal coordinates are much less sensitive to the initial Hessian data--the (default) diagonal Hessian is, in general, just as good as a full mechanics-generated Hessian. Thus, if you are optimizing in natural internal coordinates, an input Hessian is not obligatory for good performance.
Table 1
. Efficiency of optimization under various conditions
Number of optimization cycles needed to reach convergence for minimization using Cartesian and internal coordinates, starting with a unit, a diagonal, or a molecular mechanics-derived Hessian. Calculations were done with Turbomole, but the same general results would be obtained in any DFT calculation.
|
molecule
|
number of atoms
|
symmetry group
|
number of variables
|
Cartesian coordinates
|
internal coordinates
|
|---|
| unit Hessian
| mechanics Hessian
| unit Hessian
| diagonal Hessian
| mechanics Hessian
|
|---|
|
water
|
3
|
C2v
|
2
|
5
|
5
|
7
|
5
|
6
|
|
ammonia
|
4
|
C3v
|
2
|
7
|
6
|
7
|
5
|
6
|
|
acetylene
|
4
|
D h h
|
2
|
7
|
6
|
5
|
7
|
6
|
|
benzene
|
12
|
D6h
|
2
|
6
|
4
|
5
|
3
|
4
|
|
ethane
|
8
|
D3d
|
3
|
7
|
4
|
6
|
4
|
5
|
|
propadiene
|
7
|
D2d
|
3
|
10
|
5
|
8
|
5
|
5
|
|
neopentane
|
17
|
Td
|
3
|
10
|
5
|
7
|
5
|
5
|
1,3,5-trifluoro-
benzene
|
12
|
D3h
|
4
|
7
|
5
|
6
|
5
|
5
|
|
hydroxysulfane
|
4
|
C1
|
6
|
21
|
11
|
17
|
10
|
8
|
|
disilyl ether
|
9
|
C2v
|
7
|
27
|
10
|
13
|
8
|
8
|
|
acetone
|
10
|
C2v
|
8
|
22
|
7
|
7
|
6
|
6
|
|
furan
|
9
|
C2v
|
8
|
10
|
8
|
11
|
7
|
8
|
|
naphthalene
|
18
|
D2h
|
9
|
11
|
5
|
9
|
7
|
5
|
|
methylamine
|
7
|
Cs
|
10
|
10
|
5
|
7
|
5
|
6
|
1,3,5-trisilacyclo-
hexane
|
18
|
C3v
|
11
|
36
|
8
|
14
|
8
|
8
|
1,3-difluoro-
benzene
|
12
|
C2v
|
11
|
8
|
5
|
9
|
6
|
5
|
|
ethanol
|
9
|
Cs
|
13
|
18
|
6
|
8
|
6
|
6
|
|
difluoropyrazine
|
16
|
C2h
|
15
|
21
|
8
|
11
|
9
|
9
|
1,5-difluoro-
naphthalene
|
18
|
C2h
|
17
|
16
|
6
|
11
|
7
|
6
|
|
benzidine
|
26
|
D2
|
18
|
26
|
10
|
25
|
12
|
9
|
|
benzaldehyde
|
14
|
Cs
|
25
|
17
|
6
|
9
|
6
|
6
|
4-methyl-3-pen-
ten-2-one
|
17
|
Cs
|
28
|
38
|
7
|
9
|
8
|
7
|
2-amino-4-pteri-
dinol
|
17
|
Cs
|
31
|
23
|
9
|
12
|
11
|
10
|
2-hydroxybi-
cyclopentane
|
14
|
C1
|
36
|
45
|
15
|
23
|
13
|
15
|
|
caffeine
|
24
|
Cs
|
42
|
39
|
10
|
11
|
10
|
12
|
|
histidine
|
20
|
C1
|
54
|
102
|
30
|
44
|
23
|
19
|
|
dimethylpentane
|
23
|
C1
|
63
|
29
|
9
|
15
|
16
|
12
|
|
menthone
|
29
|
C1
|
81
|
100
|
14
|
26
|
16
|
13
|
However, under certain circumstances the topology of your molecule might be such that OPTIMIZE has difficulty generating a complete set of internal coordinates. If this occurs, the default procedure is to switch to Cartesian coordinates, where a starting Hessian is needed. Bear this in mind when deciding whether or not to generate a Discover Hessian for your ab initio optimization.
Optimization parameters are set within the Insight environment by selecting the Optimize/Opt_Parameters command. The commands in the Optimize pulldown in the DMol user interface appear greyed out if an OPTIMIZE calculation has not been selected (via the Setup/System command). This is intended to keep you from defining parameters that do not affect the calculation.
When the Optimize/Opt_Parameters command is first selected, the parameter block shown in Figure 2 appears. The parameters and their meanings are discussed below.
Coordinate_System refers to the coordinates used to describe the molecule to be optimized. These can be Cartesian or Internal coordinates--the latter are generated automatically by OPTIMIZE, so no user input is required. The default is Auto, which means that OPTIMIZE attempts to generate a set of internal coordinates and use these, but if this fails, the program switches to Cartesian coordinates. Specifying Cartesian or Internal forces the coordinate choice. For successful optimization using Cartesian coordinates, a reliable starting Hessian (read from a file specified by Hessian_File after toggling Starting_Hessian to on) is recommended.
Use_Constraints is toggled on (indicated by highlighting and a pushed-in appearance) if you already set any geometric constraints for your molecule with the Optimize/Constraints command. Two algorithms are available for constrained optimization: Penalty functions and Lagrange multipliers. The former is more robust; the latter is more accurate and more efficient. The default is Lagrange_Penalty, which means that OPTIMIZE first tries the Lagrange multiplier algorithm and, if this fails, it then switches to penalty functions. The other option, Penalty_Lagrange, uses penalty functions and, at convergence, refines the final structure using Lagrange multipliers. Specifying Penalty or Lagrange forces a particular algorithm.
You may have convergence problems if you use angle constraints near 0° or 180°. It may be necessary to optimize using only penalty functions first and then use the Lagrange multiplier method in a separate run after the first has converged.
The current maximum number of specific distance or angle constraints that is allowed is 100. More "constraints" can be set by freezing atom positions: any number of atoms may be frozen. You may request other distance and angle constraints and frozen coordinates in the same job; however, take care that your constraints are not mutually exclusive.
Note that constrained transition-state searches can be done only with the Lagrange multiplier method--the penalty function algorithm cannot be used.
Use_Symmetry is on if you used the Symmetry/Find_Pt_Groups command to find the symmetry of your molecule. (Symmetry is not used in the optimization unless the Symmetry/Find_Pt_Groups command has been executed, even if Use_Symmetry is on.)
Locate_Trans_State should be toggled on if you want to optimize to a transition state. The Calc_Mode parameter becomes accessible when Locate_Trans_State is on and is used to indicate which Hessian mode to maximize--the default is to maximize along the lowest mode. Other modes can be requested by setting Calc_Mode accordingly; however, this is not recommended unless you know the Hessian structure in some detail. Without a reasonable starting Hessian, transition-state searches have limited chances of success.
GDIIS_Mode invokes the GDIIS algorithm of Pulay (Pulay 1982). This is available only for minimization. GDIIS calculates the new geometry as a linear combination of the current geometry and geometries from previous optimization steps; the total number of such geometries utilized is given by Max_DIIS, which becomes accessible when GDIIS_Mode is set to Manual. A suggested range for Max_DIIS is 2-10. Setting GDIIS_Mode to Automatic causes OPTIMIZE to estimate a suitable value for Max_DIIS based on the number of degrees of freedom.
Tolerances refers to the criteria that must be met for the geometry to be considered converged. The Grad_Conv criterion and either the Ener_Conv or Disp_Conv criteria (or both) must be satisfied for the optimization to be considered converged.
Grad_Conv is the convergence criterion for the maximum component of the gradient vector (in atomic units). The default is 0.0003, which should be more than sufficient for most purposes. To obtain accurate vibrational frequencies for very floppy molecules, this value should be reduced. For general optimizations using DMol, a value of 0.001 is more suitable, due to the heavy numerics involved in this code.
Ener_Conv is the convergence criterion for the energy change from the previous optimization step (in atomic units). The default is 0.000001 Hartree. For general optimizations using DMol, a value of 0.00005 is more suitable, due to the heavy numerics involved in this code.
Disp_Conv is the convergence criterion for the maximum predicted displacement, that is, the maximum component of the displacement vector for the next step (in atomic units). The default is 0.001, which should be more than sufficient for most purposes. For very floppy molecules this value can be increased without affecting the energetics. For general optimizations using DMol, a value of 0.001 is more suitable, due to the heavy numerics involved in this code.
Cycles is the maximum number of optimization cycles allowed for the job. The default is 50, which should be more than enough under most circumstances. If convergence is not achieved in Cycles steps, the job aborts.
Max_Disp is the maximum allowed step size, that is, the maximum permitted length of the displacement vector from one optimization cycle to the next (in atomic units). The default is 0.3. For awkward optimizations (e.g., to transition states or very shallow minima) with starting geometries known (or suspected) to be close to the final converged geometry, Max_Disp should be reduced.
All optimizations, especially those using Cartesian coordinates, should be initiated with a good starting Hessian. For standard minimizations, a good Hessian can be obtained from the Discover program; for transition-state searches, DMol should be instructed to calculate a Hessian before starting the optimization (by selecting the Setup/System and choosing the Frequency option).
Toggling Hessian_Update to on enables you to access the Hessian_Update_Mode parameter, which indicates the manner in which the starting Hessian is updated during the optimization procedure. Three update methods are supported: Powell, BFGS, and BFGS_Safe. Defaults are provided, depending on the settings of various other parameters in this parameter block. These defaults should not be changed.
When the parameter block initially appears (Figure 2), the default options that are applicable to an unconstrained minimization using the EF algorithm are highlighted. Indeed, if a standard minimization is all you intend to do, there is no need to select the Optimize/Opt_Parameters command at all. The parameter block expands (or greyed-out parameters become accessible) when certain options are selected. For example, setting GDIIS_Mode to Manual makes the Max_DIIS parameter box accessible (see Figure 3), allowing the size of the iterative subspace to be set (the default is 4). At the same time, the Hessian_Update_Mode changes from BFGS (the default for EF minimization) to BFGS_Safe (the default for GDIIS).
When you have set all the parameters in this parameter block, select Execute to actually set the internal DMol parameters and to exit the parameter block.
Volumetric and print parameters
The Volumetric and Print parameters of the Setup/Parameters command allow you to set up several output-related parameters that determine the content of the .outmol file or generate plotting data. All these parameters are off by default. Molecular orbitals, eigenvalues, and occupations obtained at the end of the calculation are always output.
Densities
Set the Charge_Density parameter on to generate plot data to represent the total charge density. Set the Deformation_Dnsty parameter on to generate plot data for the deformation density (molecular charge density minus atomic charge densities). Set the Spin_Density parameter on to generate plot data for the spin density (alpha-spin minus beta-spin density).
Electrostatic potential
Set the Potential parameter on to generate plot data for the electrostatic potential.
Molecular orbitals
Set the HOMO parameter on to generate plot data for the highest occupied molecular orbital. Set the LUMO parameter on to generate plot data for the lowest unoccupied molecular orbital. Set the Other_MO parameter on so that you can enter numbers, separated by spaces, in the Molecular_Orbitals parameter box to specify which molecular orbitals to plot. List the lowest orbital energy first. (You cannot plot frozen-core orbitals.)
Optional output file contents
Set the MO_Coefficients parameter on to include molecular orbital coefficients in the output file. Set the Eigenvalues parameter on to include the eigenvalues in the output file.
Setup_Grid_Output command
The Setup/Setup_Grid_Output command allows you to set up the parameters that determine the boundaries of the grid for generation of 3D data.
The Grid_Style parameter allows you to select the method used to compute the grid boundaries. Setting Grid_Style to Enclosure automatically determines the upper and lower limits of the x, y, and z dimensions of the grid, based on the coordinates of the molecule and the value of the Border_Space parameter. The value of the Border_Space parameter (in angstroms) is added to the maximum value in each of the x, y, and z dimensions spanned by the molecule, and, similarly, subtracted from the minimum value in each dimension to define the 3D rectangular region of the grid.
You also need to specify the method for determining the positions of the grid points. With Grid_Style set to Enclosure, you specify the grid points with the Number_of_Steps parameter. The Number_of_Steps parameter allows you to choose the number of points per dimension of the 3D grid. These points are evenly spaced in each of the x, y, and z dimensions. This method provides a constant number of points in each dimension, but not a constant point density for any molecule or assembly. Note that the lower boundaries of the grid will be shifted so that the grid points fall exactly on the grid boundaries.
With Grid_Style set to Extents, you explicitly specify the upper and lower grid boundaries in each of the X, Y, and Z dimensions, using the Lower_Bounds and Upper_Bounds parameters.
By setting Grid_Style to Extents, you specify the positions of the grid points with the Grid_Step parameter. The Grid_Step parameter allows you to specify the distance, in angstroms, between points in each dimension of the 3D grid. This value must be greater than zero. This method provides a constant density of points in each dimension of any size grid.
Setting up the background job
Use of the Background_Job pulldown is optional. If it is not used, the default is to run all jobs on the local host in Cont_Insight mode (see the Background_Job/Setup_Bkgd_Job command). If you prefer this mode, you do not need to read the remainder of this subsection.
When the Background_Job/Setup_Bkgd_Job command is used, the background job list shows only those background jobs that are run from the current module and can be run on a remote host. If the module contains only one job, the parameter is automatically filled in. The list of hosts shows only those hosts that are associated with that background job in the background_job_hosts file at your site. It is possible for you to specify a remote host that is unavailable (off line, for instance) or for which you have no login account.
Use the Background_Job/Control_Background_Job command to coordinate running background jobs by detaching selected jobs from or attaching them to Insight. In addition, you can use this command to specify the interval for invoking a task specific to a particular background job for processing its output.
Every background job submitted via the generic background utility is assigned a job number. This number is displayed in the information area when the job is submitted (e.g., Starting Module background job ... as job 1). You should note the job number when the job is submitted, since it can be used later to check on the job's completion status or kill the job.
The Setup_Bkgd_Job command does not actually run the command; it simply records your host and execution mode preferences. The default host is Local. Your selected host and Execution_Mode are used for any subsequent background jobs for the duration of the Insight session. When you start up a new session, all background job parameters are set to their default values.
The Execution_Mode parameter allows you to run a background job concurrently (Cont_Insight) or interactively (Wait_For_Job) or to simply create the necessary command files to submit the job, but not actually execute them (Cmd_File_Only).
The Send_Mail parameter allows you to have the system send you an electronic mail message upon completion of the background job. This parameter is not active if Execution_Mode is set to Wait_For_Job. You may find this option useful when running long jobs where you exit the Insight program before the job completes.
The Save_Cmd_Files parameter allows you to save the command file used to submit the background job (bkgd_job_run_name#.csh). Otherwise this file is deleted when the job completes. This parameter is not active when Execution_Mode is set to Cmd_File_Only.
All background jobs return a completion status. The completion status is an integer code that indicates success, failure, and/or reason for failure of the job. The status code is always displayed when you are notified that the job has completed.
If you consistently want to send background jobs to another host, you can modify your personal Insight startup file to invoke Setup_Bkgd_Job for each module/background job(s) that you want to automatically assign. Note that you must first change to the appropriate module in which the background job's interface is found before using Setup_Bkgd_Job to set a preference for that module's background jobs.
The Completion_Window parameter can be used to prevent the notification window from appearing when the background job completes. The default value is on.
Support for the network queuing system (NQS) is now available in the Background_Job/Setup_Bkgd_Job command.
For the user interface to present parameter defaults and a value-aid containing available queues, and to correctly formulate an NQS command, the user's NQS queue environment information must be provided to the Insight program. The Background_Job_Hosts file contains the NQS queue information, or you may enter the required information directly using the Background_Job/Setup_Bkgd_Job command.
Based on the parameter values provided in the Background_Job/Setup_Bkgd_Job command for Queued Submission_Mode, the Insight background job mechanism formulates a standard NQS command and starts a process to execute it. It is assumed that the NQS command constructed by the Insight program functions with your NQS configuration.
Starting the job
Use the Run/Run_DMol command to assign a name to and initiate your DMol job. The run name also identifies the input and output files associated with the job.
Monitoring a background job
The Background_Job/Completion_Status command has three modes of operation. The One_Job option displays a brief message indicating if a specific job has completed. The message is displayed in the information area of the screen. Certain background jobs generate a status file containing additional information while they are running. If this additional status information is available, it is displayed in the textport. If All_Jobs is chosen, the job number, job name, run name, status code, and job status are displayed in the textport for every job submitted during the current Insight session. The Look_Up_Status option is used to look up the meaning of a return status code.
The Report_Mode parameter is used to indicate what information you would like the command to return: status of one job, status of all jobs, or the meaning of a return status code from a particular job.
The Job_Number parameter becomes active when One_Job is selected. It is used to specify the background job that you want to monitor.
The Background_Job and Status parameters become active when Look_Up_Status is chosen. They are used to specify a status code that you want to look up.
The Kill_Bkgd_Job command is used to stop execution of a background job by killing its process and, optionally, deleting its output files.
The Job_Number parameter is used to specify which background job to kill. A value-aid containing a list of all currently running background jobs is provided.
If the Save_Output parameter is toggled on, then all output files generated by the background job are saved when the job is killed. The default value of this parameter is off, meaning that all output files are deleted.
Visual aids to analyzing results
Displaying orbital contours
The reactivities of molecules may often be determined in terms of the sizes and detailed shapes of the molecular orbitals that comprise their valence manifolds (Hout et al. 1983). Inspection of the shapes and symmetries of only the highest occupied and lowest unoccupied molecular orbitals is often sufficient to determine whether or not two molecules can react and, if they do react, what the stereochemistry of the product is likely to be (Hout et al. 1983).
You can use the Analyze/Orbital_Contour command to automatically contour both the plus and minus phases of a molecular orbital in one step (the Grid/Contour command requires two separate steps for contouring the plus and minus phases of an orbital; and the contour levels, contour colors, and contour name root parameters have no default values.) You can also use this command to recalculate an orbital contour, by toggling the Recalc_Contours parameter on.
The name of the grid that contains the relevant data is specified with the Scalar Grid Name parameter. A name is given to a grid during its creation with the Grid/Get command (the Grid pulldown is accessed from one of the icons along the side of the main Insight window).
The Contour Name parameter specifies the root name to be given to a created contour objects or the name of the contour pair that you want to recalculate. When calculating a new contour, you usually do not need to pay any attention to this parameter, since a unique default name is derived from the value of Grid Name parameter. When recalculating, both the positive and negative contours are recalculated.
The Orbital_Amplitude parameter is used to specify the value of the contour to create. For orbital amplitude contours, the default value of is usually appropriate.
The colors assigned to the plus and minus phases of the orbital contour are specified with the Plus Contour Color and Minus Contour Color parameters, respectively. The color can be specified as a character string such as red, as an RGB triplet of integers specifying the red, green, and blue contributions, or by choosing a standard or custom-mixed color from a color palette value-aid. Control of color is explained further in the Insight II User Guide.
Displaying charges
The Analyze/Charge Display command displays or loads charges for the molecule specified by Object Name. You can display, undisplay, or load the charges, according to the setting of the Charge_Operation parameter.
The Charge_Type parameter specifies the type of charge that is being employed for the Charge_Operation parameter. If Charge_Type is set to Current_Charges, the partial charges obtained from Insight are considered. (The combination Charge_Operation equals Load and Charge_Type equals Current_Charges therefore has no effect.)
The remaining choices of the Charge_Type are the types of atomic charges that are supplied by DMol, Turbomole, or Zindo.
The color of positive and negative charges is specified through the Charge_Plus_Color and the Charge_Minus_Color parameters, respectively. The charge spheres can be scaled using the Charge_Scale_Factor parameter, which defaults to 1.0 (no scaling).
Displaying optical absorption spectra
The Analyze/Spectrum_Display command allows you to display the calculated optical absorption spectra.
The initial state of this command (Spectrum_Action = Create, File Name = *.optabs) is to take the optical absorption calculation results from the .optabs file (see Optical absorption results) and create a bar graph in the display area of the Insight window.
The graph can then be modified by setting Spectrum_Action to Modify and applying line broadening. Two modes of line broadening are supported:
- Functional form =

- Functional form =

The Sigma parameter determines the width of the function, and x is the difference between the current energy and the transition energy. The resolution of the new, broadened spectrum plot is defined by the number of Points. The energy interval for which the broadened spectrum plot is to be created is defined by its lower and upper boundaries (E_Min and E_Max).
E_Min defaults to zero if E_Min or E_Max is activated after changing the Sigma parameter. E_Max defaults to [(the highest transition energy) plus (three times Sigma)] if E_Max or E_Min is activated after changing the Sigma parameter.
Customizing the
Spectrum_Display command
So that you can display the optical absorption spectrum with energy units different from the default units of wavenumbers (cm-1) used throughout the DMol module in Insight, a mechanism is provided for customizing the energy units. The customization is based on the environment variable $SPECTRUM_UNITS. The Spectrum_Display command recognizes the following settings automatically:
SPECTRUM_UNITS cm-1 (wavenumber units, default)
SPECTRUM_UNITS eV (electron Volt units)
SPECTRUM_UNITS Ry or Rydberg (Rydberg units)
SPECTRUM_UNITS Ry or Rydberg (Rydberg units)
SPECTRUM_UNITS a.u. (atomic units)
It also recognizes arbitrary units if specified in this format:
SPECTRUM_UNITS Unit_Name Unit_Conversion_Factor
The Unit_Name is a character string that is used as the title of the x axis of the graph displaying the spectrum. The Unit_Conversion_Factor is the factor that converts from atomic units--the units that the .optabs data file refers to--into the Unit_Name units.
This setup can be written as the following BCL Macro called OA_Units, which you can add to the Analyze pulldown:
Define_Macro OA_Units \
Enumer Energy_Unit \
Short_Ident Unit_Name \
Short_Float CnvFactor
Def_enum Energy_Unit "cm-1"
Def_enum Energy_Unit eV
Def_enum Energy_Unit Rydberg
Def_enum Energy_Unit "a.u."
Def_enum Energy_Unit Specified
Set_enum Energy_Unit "cm-1"
Param_Comment Energy_Unit "Optical Absorption" Center
Param_Comment Unit_Name "Unit Specification" Center
Lstring Scr
Condition Unit_Name Energy_Unit Specified off off
Condition CnvFactor Energy_Unit Specified off off
If ( $Energy_Unit != Specified )
Env_var SPECTRUM_UNITS Set $Energy_Unit
End
Else
Scr = $Unit_Name // " " // $CnvFactor
Env_var SPECTRUM_UNITS Set $Scr
End
Env_var SPECTRUM_UNITS Get
End_Macro
DMol
Add_To_Pulldown OA_Units Analyze
(See the Insight documentation for information on adding macros to pulldowns.)
Specifying isotopes
The default parameter block for the Isotope_Setup command looks like:

The Isotope_Setup command provides basic capability for specifying particular atomic masses before performing a normal mode analysis (below). It also provides access to default isotopic masses for a wide range of nuclear isotopes. The information is taken from a separate data file and can therefore be customized.
The Isotope parameter defines the action to be taken. The Standard_Weight and Substitute_Isotope settings of the Isotope parameter define the atomic mass for all atoms specified with the Atom parameter. The Atom parameter's scope is limited to atoms of the same element type.
The Substitute_Isotope setting makes two additional parameters accessible: Isotope Name and Isotope Mass.
Isotope Name refers to the name of the isotope that is obtained by prefixing the number of nucleons to the element symbol. The information is read from the q_isotopes.dat data file (see Files). The isotopes for a given element are listed in the Isotope List value-aid. For example, for hydrogen you would find the entries H, 1H, 2H, and 3H. The entry with only the element symbol (H here) refers to the standard weight, which is an average of isotope masses according to their relative abundances. The Isotope Mass parameter is entered automatically, depending on what Isotope Name is selected, but it can be modified. The Isotope Name is resolved to the first element found if the Atom parameter contains a wildcard.
The List_Current setting activates two parameters that are related to visualizing the current atomic mass definitions: List_In_Table and Label_Atoms.
The List_In_Table parameter, if toggled on, writes all the atomic masses, along with the atom names, to a table in a separate window when the command is executed. Clicking a table cell in that table adds an atomic mass label to the atom picked. This has the side effect that once the molecule is moved, labels are redrawn with the value of the partial charges.
The Label_Atoms parameter, if toggled on, attaches labels in the form of textual user objects to all the atoms in a molecule specified by the Molecule Name parameter.
The labels' styles can be modified--the relevant parameter names in the Isotope_Setup parameter block are self-explanatory--as can its precision (through the Precision parameter in the Insight Viewer's Session/Environment command).
Displaying normal mode vibrations
The Analyze/Normal_Mode command creates a graph of normal mode frequencies and intensities from an .outmol file generated in a frequency calculation. This command also shows animated normal modes (selected by picking a frequency in the graph) and allows computation of isotope effects.
The Isotope_Subst parameter appears if the harmonic frequencies are actually calculated from a Hessian matrix supplied with the File_Contains parameter set to Hessian_Matrix. If the Isotope_Subst parameter is toggled on, the values for the atomic masses specified through the Isotope_Setup command (above) are used in the normal mode computation.
The Analyze/Normal_Mode command allows you to specify a wavenumber or an energy range for which to plot the frequency spectrum. The spectrum can be broadened. Modes can be displayed as arrows, and a distort function allows you to displace the geometry in the direction of a particular mode. A table containing the normal mode frequencies is generated, which can be used for selecting modes for the various visual representations.
Statistical thermodynamics
The Analyze/Stat_Mech command is used to obtain statistical thermodynamics data from the results of a quantum-code frequency calculation or a Discover .hessian file. The default parameter block looks like:

The Analyze/Stat_Mech command operates on the basis of a frequency table supplied by the Analyze/Normal_Mode command. This frequency table is generated through a .hessian file supplied by either Discover or quantum codes such as DMol or Turbomole. The frequency table contains all the information required to perform the thermodynamic analysis, i.e., the values of all frequencies for the given system as a result of the normal mode analysis performed with the Normal_Mode command, as well as the atomic coordinates and masses needed for computing the translational and rotational contributions to the thermodynamic quantities. The atomic masses can be modified with the Analyze/Isotope_Setup command before performing the normal mode analysis.
Files needed and produced
When the Stat_Mech command is executed, it launches a background job named stat_mech, which uses one input file called stat_mech.input and produces two output files, stat_mech.output and stat_mech.tbl:
The frequency table is specified by the NMA_Frequency_Table parameter. The program checks whether this table actually contains all necessary information, since the associated Objects value-aid includes all tables that were generated in the current Insight session.
Temperature range
The range of temperatures for which thermodynamic quantities are to be computed is specified through three parameters:
The temperatures are specified as absolute temperatures in Kelvin.
Thermodynamic quantities
The thermodynamic quantities computed are:
For each of these four quantities, the contributions from translational, rotational, and vibrational motions can be written to a .tbl file. Exactly which thermodynamic data are written to the .tbl file depends upon the settings of parameters in the Stat_Mech command. For example, the S_Vibration parameter controls whether the vibrational contribution to the entropy is written.
Additional parameters control the printing of the zero-point vibrational energy (Report_ZPVE) and of the moments of inertia (Report_Rotor_Info) to the .output file. The Report_Symmetry parameter, if toggled on, both reports the symmetry number for the particular system and includes it in the rotational partition function.
Displaying density-of-states information
The Analyze/Density_of_States command creates a total density-of-states plot by broadening the eigenvalues that are read from an .outmol or .dos file. Partial density-of-states information (e.g., contributions by atom, atom type, or atomic orbital) can be read only from a .dos file.
Once a converged electron density has been calculated, there are several ways to analyze the results, in particular the wavefunction. A convenient way of displaying the molecular orbital spectrum is by constructing and plotting the density of states. For molecular systems, this is commonly done by graphing the molecular orbitals as a function of the MO eigenvalues. The degeneracy of the orbitals is then indicated by the height of the functions. The theory is presented under Density of states analysis.
The DOS intensities can be convoluted with either a Gaussian or a Lorentzian broadening function, or you can use nonconvoluted delta functions. Display parameters like the broadening parameter NM_Sigma and the energy range Freq Min, Freq Max for the DOS graphs can be changed. The data from which the graphs are created are stored in a table. This table contains the eigenvalues, occupations and intensities, and the labels for the symmetry representations and can be used for further reference.
You can also display Loewdin or Mulliken partial density of states (PDOS) or the conventional PDOS, which is a straight projection of the atomic orbitals on the molecular wavefunctions. The advantage of Loewdin and Mulliken is that they are normalized and, for all atomic orbitals, add up to the occupation for each molecular orbital.
Displaying a summary of DMol output
The Analyze/Scan_DMol_Output command creates a short text summary (in a file called run_name.sum) of the DMol output of a job that ran or is still running. The summary includes a list of the most important keywords, the most recent geometry and gradients, the total and binding energies, and information on the molecular orbitals (including information about the symmetry, if any, and the eigenvalues).
For frequency calculations, the vibrations are included, and if properties were calculated, both Mulliken and Hirshfeld data, as well as the electrostatic moments, are summarized.
The run statistics (the computer and CPU used, total CPU time) are included at the end of the summary file. This command is also useful for polling running jobs to see how they are progressing.
Using other Insight pulldowns
The Grid pulldown (which is one of the icons along the side of the main Insight window) is used to create and manipulate an energy grid for a molecule. You can create and compute the energy grid, display and undisplay it, and write it to an output file that is readable by the Contour/Get command. You need to define the parameters in the Grid pulldown commands before plotting a DMol calculation.
Please see the Insight documentation for further information on using the Grid and other Insight pulldowns and icons.
Last updated September 25, 1997 at 03:13PM PDT.
Copyright © 1997, Molecular Simulations, Inc. All rights
reserved.

